Neuroendocrine tumors (NETs) are uncommon and can occur in the context of genetic conditions. Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant disorder of the tumor suppressor gene of the same name— MEN1, which encodes for the protein menin. Multiple endocrine neoplasia type 1 is characterized clinically by the presence of 2 or more of the following NETs: parathyroid, pituitary, and pancreaticoduodenal. 1 Pancreaticoduodenal NETs occur in 30% to 80% of patients with MEN1 and have malignant potential. Although the majority of pancreaticoduodenal NETs are nonfunctioning, patients may present with symptoms secondary to mass effect.
Genetic testing exists for MEN1, but not all genetic mutations that cause MEN1 have been discovered. Therefore, because negative genetic testing does not rule out MEN1, a diagnosis is based on tumor type and location. Neuroendocrine tumors of the biliary tree are rare, and there
are no well-accepted guidelines on how to stage them. 2-4 The following case demonstrates an unusual initial presentation of a NET in the context of MEN1.
Case Report
A 29-year-old, active-duty African-American man deployed in Kuwait presented with icterus, flank pain, and hematuria. His past medical history was significant for nephrolithiasis, and his family history was notable for hyperparathyroidism. Laboratory results showed primary hyperparathyroidism and evidence of biliary obstruction.
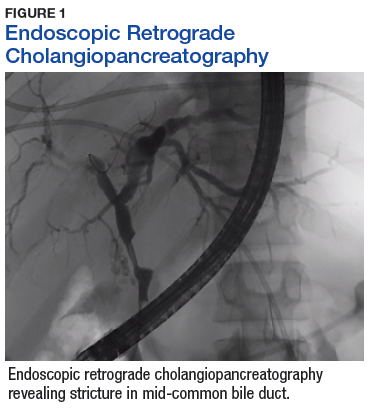
A sestamibi scan demonstrated uptake in a location corresponding with the right inferior parathyroid gland. A computed tomography (CT) scan showed nephrolithiasis and hepatic biliary ductal dilatation. Magnetic resonance cholangiopancreatography (MRCP) revealed both intra- and extrahepatic ductal dilatation, focal narrowing of the proximal common bile duct, and possible adenopathy that was concerning for cholangiocarcinoma. Endoscopic retrograde cholangiopancreatography (ERCP) demonstrated a 1 cm to 2 cm focal stricture within the mid-common bile duct with intra- and extrahepatic ductal dilatation (Figure 1). An endoscopy showed no masses in the duodenum, and anendoscopic ultrasound showed no masses in the pancreas. Endoscopic brushings and endoscopic, ultrasound-guided, fine-needle aspiration
cytology were nondiagnostic. Exploratory laparotomy revealed a dilated hepatic bile duct, an inflamed porta hepatis, and a mass involving the distal hepatic bile duct.
The patient underwent cholecystectomy, radical extra hepatic bile duct resection to the level of the hepatic bifurcation, and hepaticojejunostomy. Gross examination of the specimen showed a nodule centered in the distal common hepatic duct with an adjacent, 2-cm lymph node. The histologic examination revealed a neoplastic proliferation consisting of epithelioid cells with round nuclei and granular chromatin with amphophilic cytoplasm in a trabecular and nested architecture.
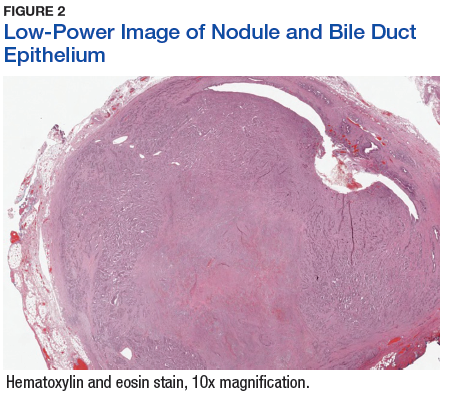
The tumor was centered in the submucosa, which is typical of gastrointestinal NETs (Figure 2). There was no evidence of direct tumor extension elsewhere. About 40% of the tumor cells contained eosinophilic, intracytoplasmic inclusions (Figure 3). The tumor did not involve the margins or lymph node.
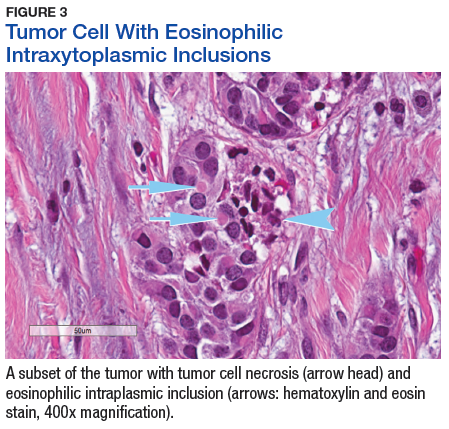
Positive staining with the neuroendocrine markers synaptophysin and chromagranin A confirmed a well-differentiated NET. The intracytoplasmic inclusions stained strongly positive for cytokeratin CAM 5.2. The tumor had higher-grade features, including tumor cell necrosis, a Ki-67 labeling index of 3%, and perineural invasion. The 2010 World Health Organization (WHO) criteria for NET of the digestive system classified this tumor as a grade 2, well-differentiated NET and as stage 1a (limited to the bile duct). 4