Postoperatively, octreotide scan with single-photon emission computed tomography (SPECT)-CT did not show additional masses or lesions. Serum pancreatic polypeptide was elevated, with the remaining serum and plasma NET markers—including gastrin, glucagon, insulin, chromogranin A, and vasoactive intestinal polypeptide (VIP)—being within reference ranges. Genetic testing (GeneDx, Inc, Gaithersburg, MD) showed an E563X nonsense mutation in the MEN1 gene, confirming a MEN1 disorder. The patient then underwent a 4-gland parathyroidectomy with reimplantation; the parathyroid glands demonstrated hyperplasia in all 4 glands.
Biochemical follow-up at 14 months showed that the serum pancreatic polypeptide had normalized. There was no evidence of pituitary orpancreatic hypersecretion. The patient developed hypoparathyroidism, requiring calcium and calcitriol supplementation. Radiographic follow-up using abdominal magnetic resonance imaging at 16 months showed no evidence of disease.
Discussion
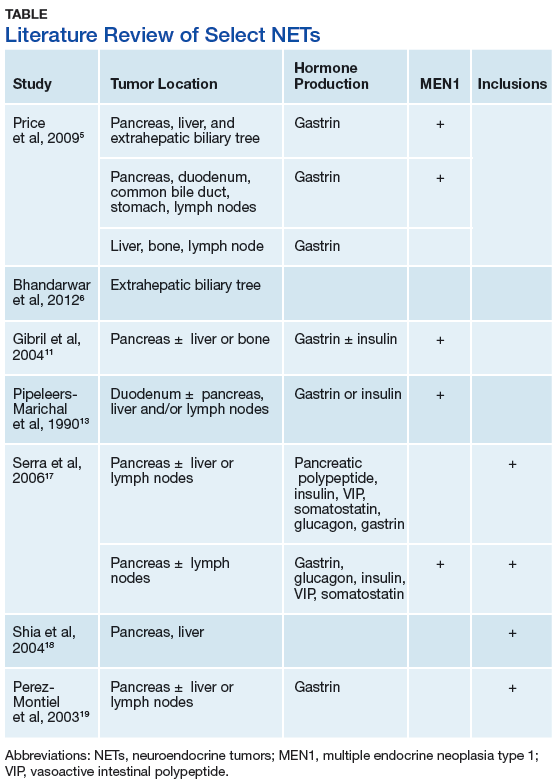
This case illustrates a genetic disease with an unusual initial presentation. Primary extrahepatic bile duct NETs are rare and have been reported previously in patients without MEN1. 5-9 Neuroendocrine tumors in the hepatic bile duct in patients with MEN1 also have been reported but only after these tumors first appeared in the pancreas or duodenum. 10 An extensive literature search revealed no prior reports extrahepatic bile duct NETs with MEN1 as the primary site or with biliary obstruction, which is why this patient’s presentation is particularly interesting. 5,6,10-13 The table summarizes select reports of NETs.
Tumor location in this patient was atypical, and genetic testing guided the management. Serum MEN1 genetic testing is indicated in patients with ≥ 2 tumors that are atypical but possibly associated with MEN1 (such as adrenal tumors, gastrinomas, and carcinoids) and in patients aged < 45 years with primary hyperparathyroidism. 14,15 The patient in this study was aged 29 years and had hyperparathyroidism and an NET of the hepatic bile duct. This condition was sufficient to warrant genetic testing, the results of which affected the patient’s subsequent parathyroid surgery. 15 Despite the suggestion of unifocal localization on the sestamibi scan, the patient underwent the more appropriate subtotal parathyroidectomy. 14 The patient’s tumor most likely originated from a germline mutation of the MEN1 gene.
As a result of the patient’s genetic test results, his daughter also was tested. She was found to have the same mutation as her father and will undergo proper tumor surveillance for MEN1. There was no personal or family history of hemangioblastomas, renal cell carcinomas, or cystadenomas, which would have prompted testing for von Hippel-Lindau disease. Likewise, there was no personal or family history of café-au-lait macules and neurofibromas, which would have prompted testing for neurofibromatosis type 1.
Due to the paucity of cases, there are currently no well-accepted guidelines on how to stage extrahepatic biliary NETs. 3-5,16 The WHO recommends staging according to adenocarcinomas of the gallbladder and bile duct. 3 As such, the pathologic stage of this tumor would be stage 1a.
The significance of the intracytoplasmic inclusion in this case is unknown. Pancreatic NETs and neuroendocrine carcinomas have demonstrated intracytoplasmic inclusions that stain positively for keratin and may indicate more aggressive tumor behavior. 17-19 In 1 report, electron microscopic examination demonstrated intermediate filaments with entrapped neurosecretory granules. 18 In a series of 84 cases of pancreatic endocrine tumors, 14 had intracytoplasmic inclusions; of these, 5 had MEN1. 17 In the present case, the patient continues to show no evidence of tumor recurrence at 16 months after resection.








