Discussion
Initial presentation of SSc can occur along a spectrum of its pathophysiology. A more severe presentation, like the one seen in Mr. P, seems to occur more frequently in African American patients relative to white patients.3 Differentiating between the 2 types—diffuse vs limited SSc—is vital to managing patients and disease progression. Limited-type SSc is more common (60%) and less severe with slower progression than is diffuse SSc.

Diffuse-type SSc (35%) includes features such as skin thickening and tightening, ILD, SRC, tendon friction rubs (palpable crepitus over tendons), and skin pigment changes.4 The specific involvement of the renal and cardiopulmonary systems accounts for the higher mortality rates in the diffuse-type.5
Many patients with SSc require periodic hospitalizations throughout their life for the acute complications of the disease. Hospitalized patients often range from age 45 to 64 years and are more often female. However, of hospitalized patients with SSc, in-hospital death rates are higher among men.3,6 Although these rates have decreased as the pathogenesis of SSc has become better understood, it is important to note that in-hospital mortality in 1995 for all patients with SSc was 7.1% and mean length of stay was 7.5 days, and in 2002 to 2003, 6.3% and 6.6 days, respectively.3 Though the burden of this disease has decreased, mortality and hospitalizations continue to persist at high rates. Understanding the pathogenesis, progression, and treatments of SSc are essential to aiding patients with this diagnosis.
Skin Involvement
A common finding and presentation for patients with SSc is related to skin involvement. Common patient complaints and exam findings include calcinosis along extensor tendons and digits, Raynaud phenomenon (seen in more than 95% of patients), sclerodactyly, telangiectasias, hyper/hypopigmentation, and pruritus.4 These findings are useful in diagnosing and monitoring patients for disease progression.
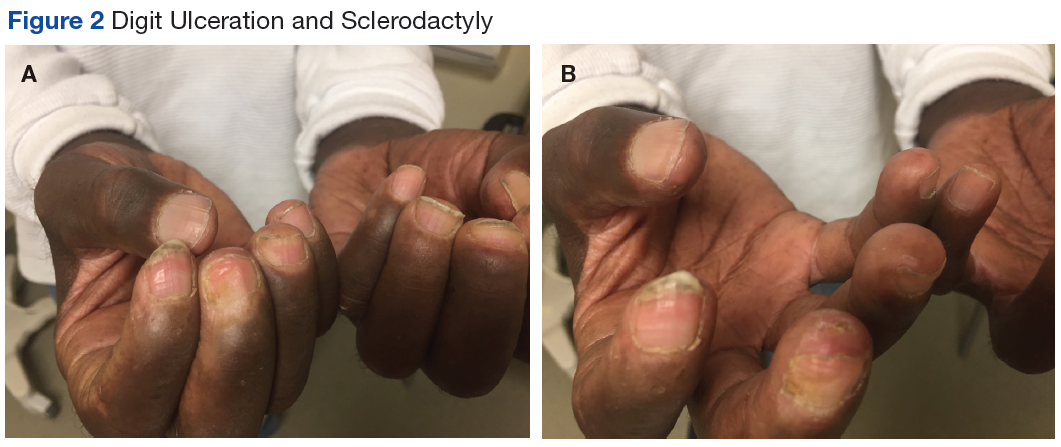
Many of the listed skin manifestations affect patients’ quality of life (QOL) but are not directly associated with mortality. However, a common and feared complication includes skin ulcers and osteomyelitis, seen in 48% and 7.7% of patients, respectively.7 Digit ulcers, areas with loss of dermis and epidermis distal to the proximal interphalangeal joints (Figures 2A and 2B), are significant because they parallel a more rapid progression of internal organ involvement.8
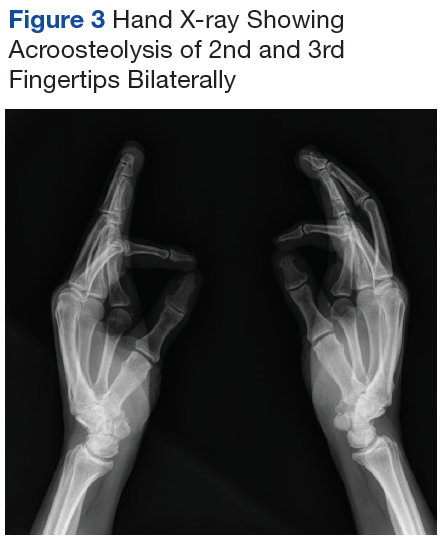
Mr. P required multiple hospitalizations and antibiotic regimens for painful digit ulcers complicated by osteomyelitis (Figure 3).
Treatment usually is aimed at infections that complicate these skin ulcers and are based on site-specific cultures. Preventive measures are aimed at the risk factors associated with digit ulcers, including decreased whole-body warmth, direct trauma to digits, smoking, and vasoconstrictors (eg, cocaine, sympathomimetics).8 Some patients may prevent ulcers by using D-penicillamine, mycophenolate mofetil, and cyclophosphamide, although definitive treatment has not been found.4 Calcium channel blockers, PDE-5 inhibitors, endothelin receptor antagonists, and prostacyclin analogues also have been used to reduce the severity of Raynaud phenomenon attacks and to decrease the number of digital ulcers (in addition to their beneficial effects on pulmonary involvement).8 Additional pharmacologic agents that have been linked to an improvement in Raynaud phenomenon and digital ulcers include statins, angiotensin-converting enzyme (ACE) inhibitors, angiotensin receptor blockers, intravenous N-acetylcysteine, vitamin E gel, and surgical options (eg, revascularization and sympathectomy).8
Pulmonary Involvement
Systemic sclerosis can lead to 2 complications in the lungs, both present in Mr. P: PAH (mean pulmonary arterial pressure > 25 mm Hg) and ILD, which together make lung involvement the leading cause of death for these patients. Limited-type SSc usually is restricted to PAH, whereas diffuse-type SSc usually leads to ILD.4
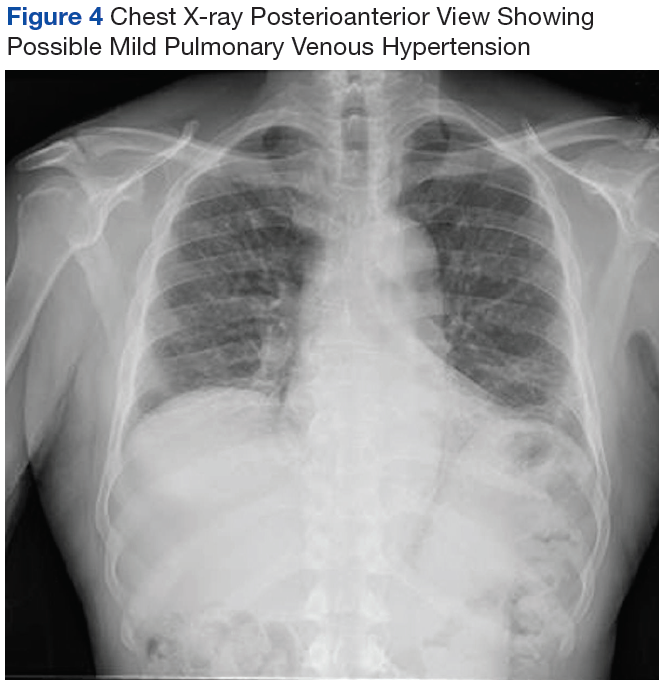
On diagnosis of SSc, an HRCT is indicated to assess the degree of lung involvement. Mr. P’s HRCT showed a 3-cm main pulmonary artery, suggestive of PAH, in addition to evidence of ILD seen on the chest X-ray (Figure 4).
Pulmonary arterial hypertension is a common finding in patients with SSc and carries a severe prognosis. Risk factors for the development of PAH, when not present on initial diagnosis, include limited-type SSc; late age of onset; Raynaud phenomenon; decreased DLCO, FVC/DLCO < 1.6; increased N-terminal pro-B-type natriuretic peptide (NT-proBNP) serum levels; and the presence of antibodies.9 Patients with both SSc and PAH have a 50% to 87% 1-year survival, whereas patients with idiopathic PAH without connective tissue disease have an 88% 1-year survival.9
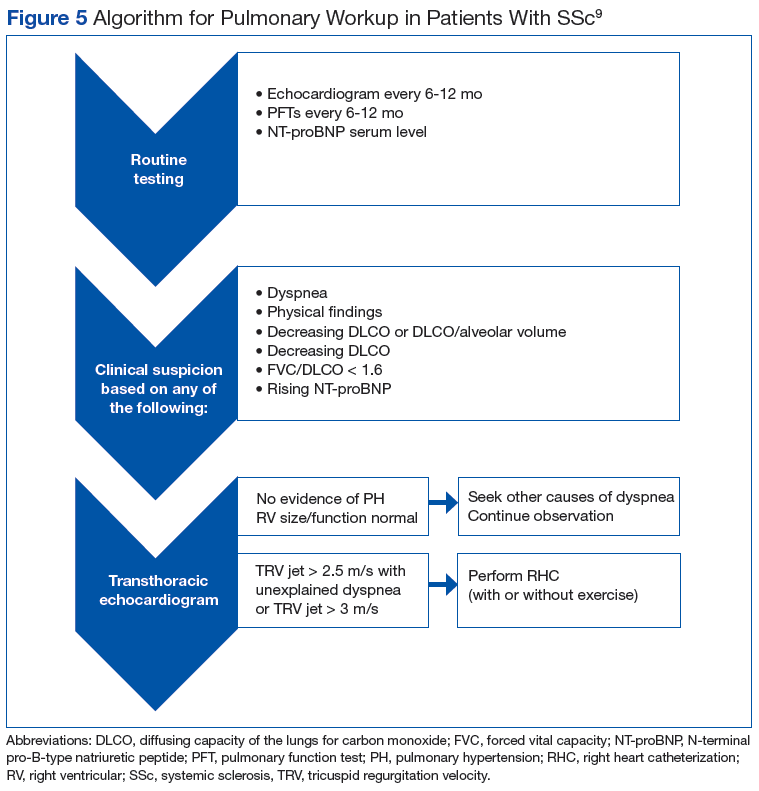
Many patients with lung involvement are asymptomatic, but some have findings of crackles and interstitial thickening on chest X-ray that can progress to cyanosis and right heart failure (cor pulmonale).10 On discovery of lung involvement (Figure 5), it is important to follow up with a PFT, primarily because the outcomes and prognosis for patients with SSc are correlated with the presenting severity of ILD and the subsequent progression of their DLCO.4,10
Treatment is directed at PAH and ILD separately. For PAH, a PDE-5 inhibitor, such as tadalafil or sildenafil, and an endothelin-1 receptor antagonist, such as ambrisentan or bosentan, are indicated. Other PAH treatments include diuretics and prostacyclin analogues (eg, epoprostenol, treprostinil, or iloprost) in addition to warfarin if patients have a history of thrombotic events.4 Mr. P’s pulmonologists deferred treatment with prostacyclin analogues given the potential for adverse effects with endothelin-1 receptor inhibitors, PDE-5 inhibitors, and calcium-channel blockers, although combination studies with bosentan and inhaled iloprost have shown promise.11
Like the skin manifestations of SSc, ILD lacks definitive treatment to prevent disease progression. However, some patients benefit from cyclophosphamide, followed by mycophenolate mofetil—which is usually better tolerated—azathioprine, haematopoietic stem cell transplant as rescue therapy, and lung transplant for life-saving treatment.10