A massive collaborative project spanning four continents and 744 research centers has revealed driver mutations in both protein-coding and noncoding regions of 38 cancer types.
 Pan-Cancer Analysis of Whole Genomes
Pan-Cancer Analysis of Whole Genomes
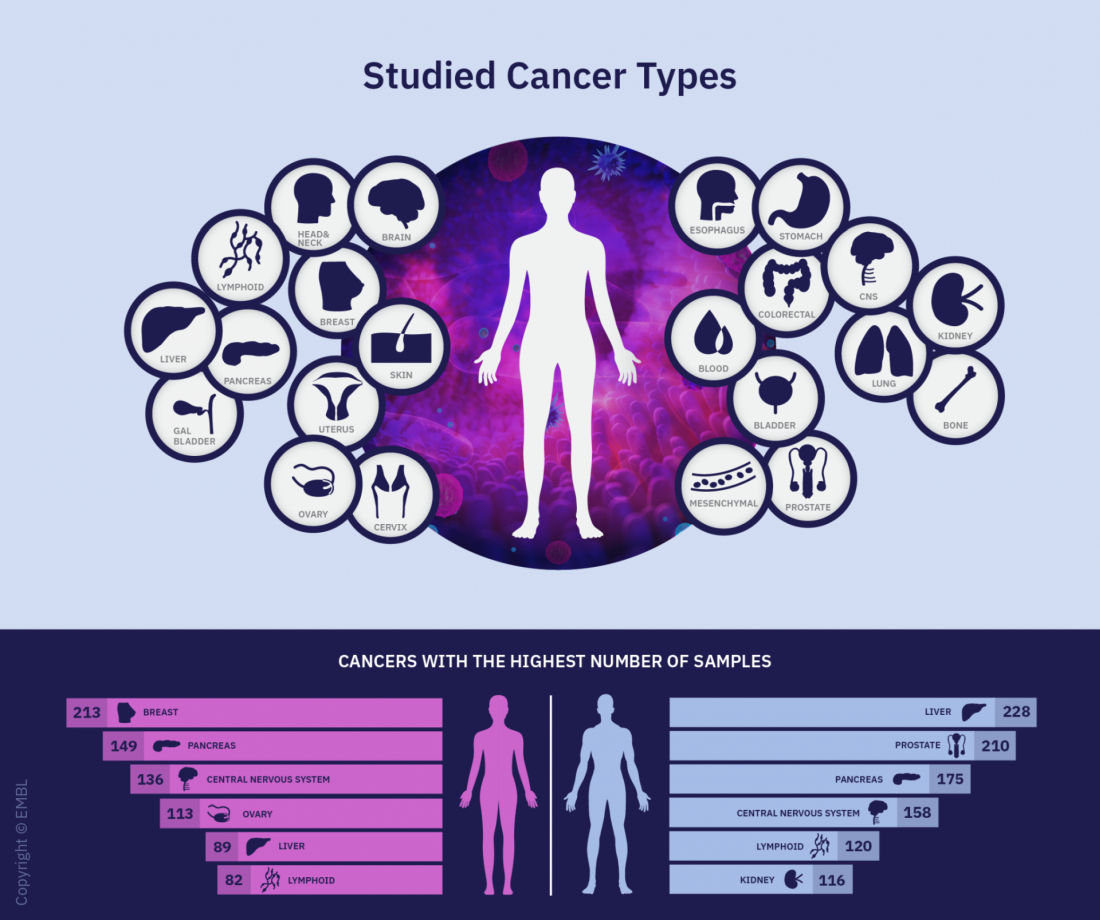
The Pan-Cancer Analysis of Whole Genomes Consortium analyzed more than 2,600 tumor samples from patients with 38 cancer types.
The Pan-Cancer Analysis of Whole Genomes (PCAWG) is an integrative analysis of the whole-genome sequences from 2,658 donors across 38 common tumor types. The findings are expected to add exponentially to what’s currently known about the complex genetics of cancer, and they point to possible strategies for improving cancer prevention, diagnosis, and care.
Six articles summarizing the findings are presented in a series of papers in Nature, and 16 more appear in affiliated publications.
“It’s humbling that it was only 14 years ago that the genomics community sequenced its very first cancer exome, and it was able to identify mutations within the roughly 20,000 protein-coding genes in the human cell,” investigator Lincoln Stein, MD, PhD, of the Ontario Institute for Cancer Research in Toronto, said in a telephone briefing.
Exome sequencing, however, covers only protein-coding genomic regions, which constitute only about 1% of the entire genome, “so assembling an accurate portrait of the cancer genome using just the exome data is like trying to put together a 100,000-piece jigsaw puzzle when you’re missing 99% of the pieces and there’s no puzzle box with a completed picture to guide you,” Dr. Stein said.
Members of the PCAWG from centers in North America, Europe, Asia, and Australia screened 2,658 whole-cancer genomes and matched samples of noncancerous tissues from the same individuals, along with 1,188 transcriptomes cataloging the sequences and expression of RNA transcripts in a given tumor. The 6-year project netted more than 800 terabytes of genomic data, roughly equivalent to the digital holdings of the U.S. Library of Congress multiplied by 11.
The findings are summarized in papers focusing on cancer drivers, noncoding changes, mutational signatures, structural variants, cancer evolution over time, and RNA alterations.
Driver mutations
Investigators found that the average cancer genome contains four or five driver mutations located in both coding and noncoding regions. They also found, however, that in approximately 5% of cases no driver mutations could be identified.
A substantial proportion of tumors displayed “hallmarks of genomic catastrophes.” About 22% of tumors exhibited chromothripsis, a mutational process marked by hundreds or even thousands of clustered chromosomal rearrangements. About 18% showed chromoplexy, which is characterized by scattering and rearrangement of multiple strands of DNA from one or more chromosomes.
Analyzing driver point mutations and structural variants in noncoding regions, the investigators found the usual suspects – previously reported culprits – as well as novel candidates.
For example, they identified point mutations in the five prime region of the tumor suppressor gene TP53 and the three prime untranslated regions of NFKBIZ (a nuclear factor kappa B inhibitor) and TOB1 (an antiproliferative protein), focal deletion in BRD4 (a transcriptional and epigenetic regulator), and rearrangements in chromosomal loci in members of the AKR1C family of enzymes thought to play a role in disease progression.
In addition, investigators identified mutations in noncoding regions of TERT, a telomerase gene. These mutations result in ramped-up expression of telomerase, which in turn promotes uncontrollable division of tumor cells.