Early Parkinsonism: Distinguishing Idiopathic Parkinson’s Disease from Other Syndromes
Journal of Clinical Outcomes Management. 2015 June;22(6)
References
Multiple system atrophy (MSA), which presents with autonomic failure in combination with motor symptoms, often poses a diagnostic challenge due to dramatic phenotypic variability. Two clinical phenotypes are recognized: MSA-C exhibits predominantly cerebellar exam features and MSA-P exhibits predominantly parkinsonian exam features and is therefore more likely to be confused with early IPD [28]. MSA-P patients can have a mild early response to dopaminergic therapy and commonly have a symmetric onset of parkinsonian features (in contrast to the asymmetry that is a hallmark of IPD). A diagnosis of probable MSA requires urinary incontinence or an orthostatic decrease in blood pressure within 3 minutes of standing by at least 30 mm Hg systolic or 15 mm Hg diastolic in addition to the motor symptoms [29]. If the autonomic dysfunction does not meet this requirement, a diagnosis of possible MSA can be made if there is at least 1 of the additional clinical or neuroimaging features ( Table 2 ). Additional supporting clinical features include orofacial dystonia, disproportionate antecollis (forward flexion of neck), camptocormia (forward flexion of the spine) or Pisa syndrome (flexion of the body and head to one side), contractures of the hands or feet, inspiratory sighs, severe dysphonia, severe dysarthria, new or increased snoring, cold hands and feet, pathologic laughter or crying, and a jerky myoclonic postural/action tremor [29]. Aside from atrophy in the brain regions listed in Table 2, typical MSA brain MRI findings include T2 hyperintensities and degeneration in the pontocerebellar tracts creating a “hot cross bun sign” in the pons. MSA-P patients have also been reported to have a finding of a hyperintense putaminal rim on T2 weighted images [30]. The reader should note that dementia is not a characteristic feature of MSA.
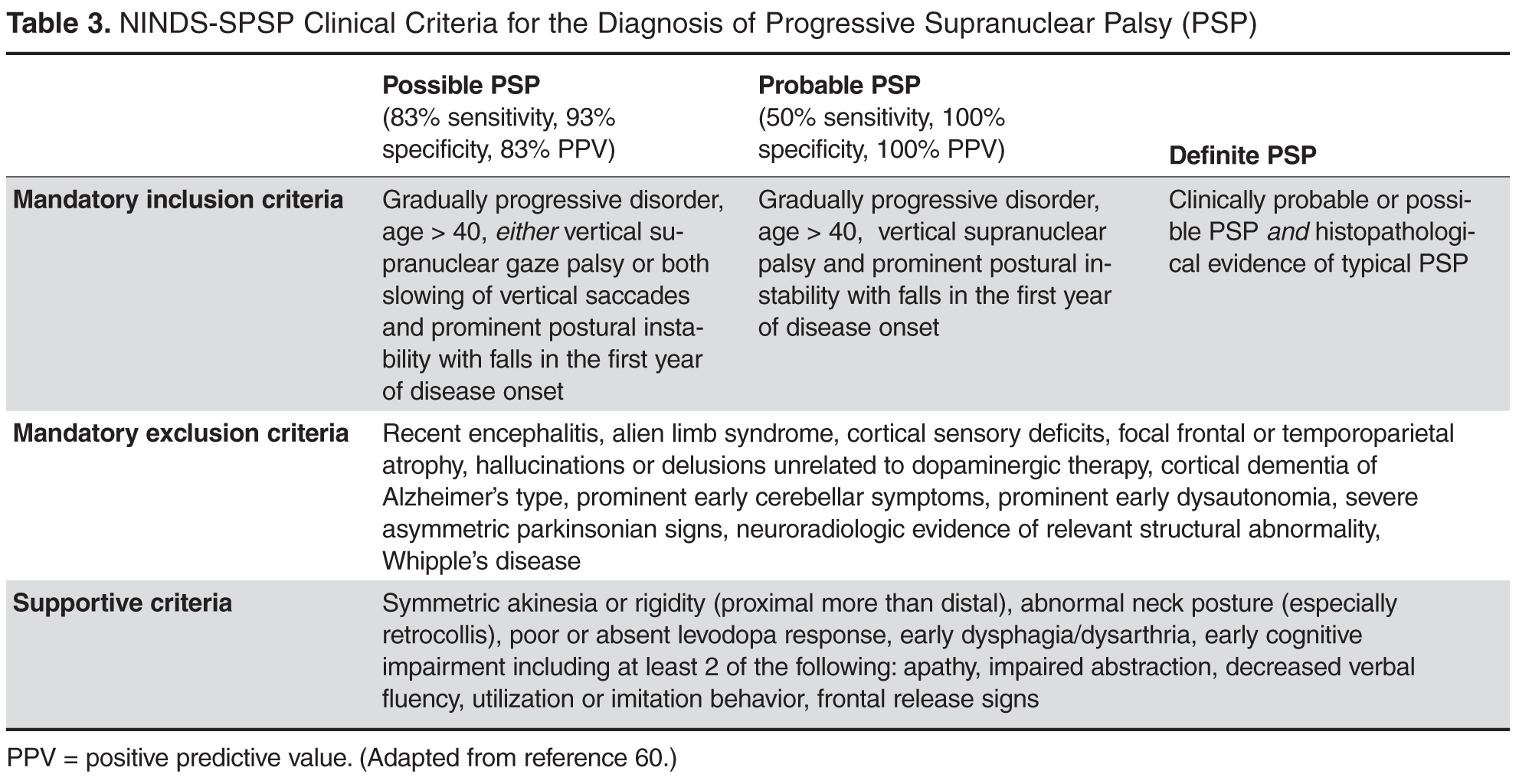
Progressive supranuclear palsy (PSP) is Parkinson-plus syndrome that often presents with parkinsonian motor signs. Some patients report an early response to dopaminergic medications, though this is typically not sustained. Other significant signs such as supranuclear vertical gaze palsy (especially in downward gaze), postural instability with repeated falls as well as frontal dementia develop early on in this condition and help to distinguish it from IPD. Gait disturbance and falls have been reported to be the presenting symptom in 90% and 62% (respectively) of PSP patients, versus IPD with gait disturbance as the presenting symptom in only 11% of patients [31,32]. Swallowing and speech difficulties are more common and more severe in PSP as well. PSP patients also typically have a symmetric onset of parkinsonian features versus the asymmetry found in most early IPD patients. Clinical criteria for the diagnosis of PSP are featured in Table 3 . Characteristic MRI findings in PSP include midbrain atrophy (reduction of antero-posterior midline midbrain diameter in axial images as well as thinning of cerebral peduncles, giving a “mickey mouse” appearance) as well as flattening or concave outline to the superior aspect of the midbrain on sagittal imaging, giving a “hummingbird sign” (normally would have an upward convex outline) [33].
Corticobasal degeneration (CBD) is more rare than the previously described Parkinson-plus syndromes. CBD typically presents with a markedly unilateral/asymmetric motor features and can mimic early IPD, but other defining features include cortical signs of progressive unilateral apraxia, limb dystonia and visual-tactile neglect (“alien limb” sign) that can lead to loss of voluntary control of the extremity. This sign has been reported in approximately half of all patients with CBD [34]. As the disease progresses, cognitive decline, dementia, dysarthria, postural instability and gait dysfunction can all occur [35]. Patients with CBD typically do not show any response to dopaminergic therapy. CBD brain MRI findings include asymmetric cortical atrophy (most commonly in the superior parietal region), bilateral basal ganglia atrophy, corpus callosum atrophy and T2 hyperintensities of the subcortical white matter and posterolateral putamen [36]. In recently published consensus criteria, Armstrong et al broadened the clinical phenotype associated with CBD to acknowledge the spectrum and overlapping phenotypes of tau-related neurodegenerative diseases [37]. The criteria for probable corticobasal syndrome require asymmetric presentation of 2 of: (a) limb rigidity or akinesia, (b) limb dystonia, (c) limb myoclonus plus 2 of: (d) orobuccal or limb apraxia, (e) cortical sensory deficit, (f) alien limb phenomena (more than simple levitation). Possible corticobasal syndrome may be symmetric and requires 1 of: (a) limb rigidity or akinesia, (b) limb dystonia, (c) limb myoclonus plus 1 of: (d) orobuccal or limb apraxia, (e) cortical sensory deficit, (f) alien limb phenomena (more than simple levitation). Unfortunately, these new criteria have not improved the specificity of diagnosis compared to previous criteria as shown by a recent longitudinal clinical and neuropathological study that found that all of their patients with a cortiocobasal syndrome but without corticobasal pathology had all met the new diagnostic criteria for possible or probable CBD [38]. The reader should be aware that Armstrong et al acknowledged that memory dysfunction is common in CBD, although this was not incorporated into the diagnostic criteria.