To the Editor:
Benign cephalic histiocytosis (BCH) falls into the group of non–Langerhans cell histiocytosis (non-LCH), which is characterized by a benign course and tendency toward spontaneous remission. Apart from BCH, the main types of non-LCH include juvenile xanthogranuloma, generalized eruptive histiocytoma, and xanthoma disseminatum.1
Benign cephalic histiocytosis is a rare form of cutaneous histiocytosis in young children. It presents as a papular eruption on the head and has not been associated with internal organ involvement.2-4 It was described in 1971 by Gianotti et al5 and was named infantile histiocytosis with intracytoplasmic worm-like bodies because electron microscopy revealed histiocytes with large cytoplasmatic inclusions composed of wormlike membranous profiles and absence of Birbeck granules. In BCH, skin lesions are located on the head including the face and sometimes on the neck. Lesions occasionally may appear on the trunk, buttocks, and thighs. Mucous membranes are not involved. The onset of disease is typical in the first year of life; however, the disease may begin within the first 3 years of life. An eruption is characterized by small, 2- to 8-mm, discrete, asymptomatic, tan to red-brown macules and papules. The lesions may persist for several months or years and subsequently flatten, becoming hyperpigmented briefly. They often completely resolve with time. Most children are otherwise healthy and developmentally normal6-9; however, diabetes insipidus has been reported in some children with BCH.10
Histologic examination of skin samples reveals the infiltrate of histiocytes, which closely approaches the epidermis, accompanied by scattered lymphocytes and a few eosinophils.1,2,11 The histiocytes express a typical macrophage marker CD68, whereas immunostaining for Langerhans cell markers such as CD1a and S-100 is negative.3,9,12,13
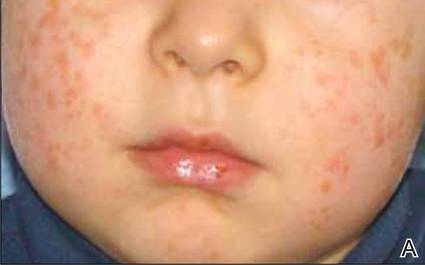
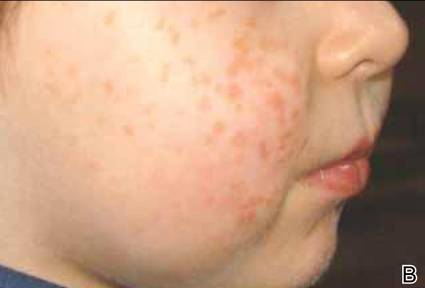
A 2.5-year-old boy was admitted to our dermatology department with suspected cutaneous mastocytosis (CM). Since the age of 13 months, he had developed small, 4- to 8-mm, dark pinkish macules and papules localized on the cheeks (Figure 1). Physical examination performed in our center revealed yellowish macules and flat papules limited to the cheeks. Darier sign was negative. The boy was otherwise healthy and developmentally normal. All laboratory tests were within reference range and his family history was uneventful.
Figure 1. Maculopapular eruption of benign cephalic histiocytosis on the cheeks (A and B). |
Histopathologic examination of the skin sample revealed a normotypic epidermis and scattered subepidermal infiltrates in the upper dermis. The infiltrates were composed of predominating histiocytes and a few mast cells and eosinophils (Figure 2). The histiocytes were slightly pleomorphic and had abundant clear cytoplasm, vesicular nuclei, and prominent nucleoli. Mitoses were absent in these cells. The majority of cells within the infiltrate expressed CD68 and were CD1a- and S-100-. However, occasional CD1a+ cells were seen. Immunostaining for mast cell marker CD117 was negative. Cutaneous mastocytosis was excluded and non-LCH was recognized. Based on the typical location, morphology, and immunophenotype of skin lesions, BCH was diagnosed. At 24-month follow-up, spontaneous regression of skin lesions was observed.
Gianotti et al7 described BCH as a separate entity of the non-LCH group of disorders and established its diagnostic criteria: (1) onset of disease within the first 3 years of life; (2) location of skin lesions on the scalp and lack of lesions on the hands, feet, mucous membranes, and internal organ involvement; (3) spontaneous complete remission of symptoms; and (4) monomorphic infiltration of histiocytes that do not express S-100 and CD1a.
The macular and flat, papular, pink-yellow or orange lesions visible on the face of our patient are characteristic of BCH. Moreover, the cheeks are the most typical location of a BCH eruption, as noted in our patient.6,7,12 The presence of histiocytic infiltrates composed of CD68+ cells strongly support the diagnosis.3,4,9-13 In contrast to other reports, occasional CD1a+ cells of Langerhans phenotype were found in our case.3,9,11,12 The proliferation of Langerhans cells in the skin and internal organs and presence of langerin are characteristic of Langerhans cell histiocytosis (LCH).1,4,14 The presence of a few CD1a cells in cases with clinical features compatible with non-LCH may suggest that some of these cases may represent a papular self-healing variant of LCH or may indicate that there is an overlap among the histiocytic syndromes (eg, non-LCH and LCH). Furthermore, many of benign histiocytic lesions may evolve over the course of time into the others.12,13 Differential diagnosis of BCH should include other benign forms of cutaneous histiocytosis, particularly the small nodular variant of juvenile xanthogranuloma and generalized eruptive histiocytoma (GEH). Juvenile xanthogranuloma pre-sents as disseminated, yellowish, nodular lesions and may be associated with ocular involvement, whereas GEH is characterized by rapid onset of the disease and disseminated nodular eruption.1,4