Biosimilars for Psoriasis
In April 2016, an infliximab biosimilar (Inflectra) became the second biosimilar approved by the FDA.4 Inflectra was studied in clinical trials for patients with ankylosing spondylitis17 and rheumatoid arthritis,18 and in both trials the biosimilar was found to have similar efficacy and safety profiles to that of the reference product. In August 2016, an etanercept biosimilar (Erelzi) was approved,5 and in September 2016, an adalimumab biosimilar (Amjevita) was approved.6
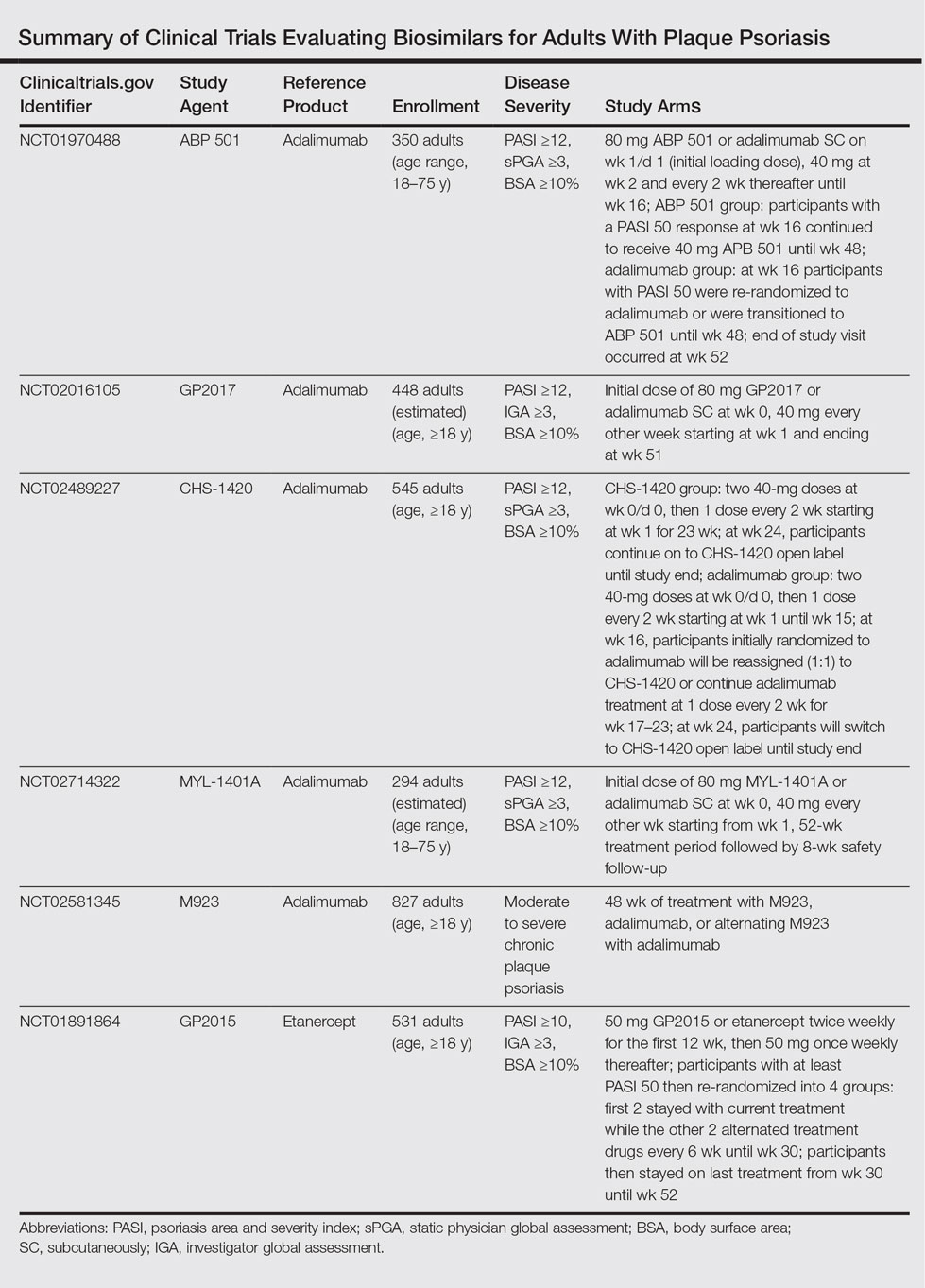
The Table summarizes clinical trials (both completed and ongoing) evaluating biosimilars in adults with plaque psoriasis; thus far, there are 2464 participants enrolled across 5 different studies of adalimumab biosimilars (registered at www.clinicaltrials.gov with the identifiers NCT01970488, NCT02016105, NCT02489227, NCT02714322, NCT02581345) and 531 participants in an etanercept biosimilar study (NCT01891864).
A phase 3 double-blind study compared adalimumab to an adalimumab biosimilar (ABP 501) in 350 adults with plaque psoriasis (NCT01970488). Participants received an initial loading dose of adalimumab (n=175) or ABP 501 (n=175) 80 mg subcutaneously on week 1/day 1, followed by 40 mg at week 2 every 2 weeks thereafter. At week 16, participants with psoriasis area and severity index (PASI) 50 or greater remained in the study for up to 52 weeks; those who were receiving adalimumab were re-randomized to receive either ABP 501 or adalimumab. Participants receiving ABP 501 continued to receive the biosimilar. The mean PASI improvement at weeks 16, 32, and 50 was 86.6, 87.6, and 87.2, respectively, in the ABP 501/ABP 501 group (A/A) compared to 88.0, 88.2, and 88.1, respectively, in the adalimumab/adalimumab group (B/B).19 Autoantibodies developed in 68.4% of participants in the A/A group compared to 74.7% in the B/B group. The incidence of treatment-emergent adverse events (TEAEs) was 86.2% in the A/A group and 78.5% in the B/B group. The most common TEAEs were nasopharyngitis, headache, and upper respiratory tract infection. The incidence of serious TEAEs was 4.6% in the A/A group compared to 5.1% in the B/B group. Overall, the efficacy, safety, and immunogenicity of the adalimumab biosimilar was comparable to the reference product.19
A second phase 3 trial (ADACCESS) evaluated the adalimumab biosimilar GP2017 (NCT02016105). Participants received an initial dose of 80 mg subcutaneously of either GP2017 or adalimumab at week 0, followed by 40 mg every other week starting at week 1 and ending at week 51. The study has been completed but results are not yet available.
The third trial is evaluating the adalimumab biosimilar CHS-1420 (NCT02489227). Participants in the experimental arm receive two 40-mg doses of CHS-1420 at week 0/day 0, and then 1 dose every 2 weeks from week 1 for 23 weeks. At week 24, participants continue with an open-label study. Participants in the adalimumab group receive two 40-mg doses at week 0/day 0, and then 1 dose every 2 weeks from week 1 to week 15. At week 16, participants will be re-randomized (1:1) to continue adalimumab or start CHS-1420 at one 40-mg dose every 2 weeks during weeks 17 to 23. At week 24, participants will switch to CHS-1420 open label until the end of the study. Study results are not yet available; the study is ongoing but not recruiting.
The fourth ongoing trial is evaluating the adalimumab biosimilar MYL-1401A (NCT02714322). Participants receive an initial dose of 80 mg subcutaneously of either MYL-1401A or adalimumab (2:1), followed by 40 mg every other week starting 1 week after the initial dose. After the 52-week treatment period, there is an 8-week safety follow-up period. Study results are not yet available; the study is ongoing but not recruiting.
A fifth adalimumab biosimilar, M923, also is currently being tested in clinical trials (NCT02581345). Participants receive either M923, adalimumab, or alternate between the 2 agents. Although the study is still ongoing, data released from the manufacturer state that the proportion of participants who achieved PASI 75 after 16 weeks of treatment was equivalent in the 2 treatment groups. The proportion of participants who achieved PASI 90, as well as the type, frequency, and severity of adverse events, also were comparable.20
The EGALITY trial, completed in March 2015, compared the etanercept biosimilar GP2015 to etanercept over a 52-week period (NCT01891864). Participants received either GP2015 or etanercept 50 mg twice weekly for the first 12 weeks. Participants with at least PASI 50 were then re-randomized into 4 groups: the first 2 groups stayed with their current treatments while the other 2 groups alternated treatments every 6 weeks until week 30. Participants then stayed on their last treatment from week 30 to week 52. The adjusted PASI 75 response rate at week 12 was 73.4% in the group receiving GP2015 and 75.7% in the group receiving etanercept.21 The percentage change in PASI score at all time points was found to be comparable from baseline until week 52. Importantly, the incidence of TEAEs up to week 52 was comparable and no new safety issues were reported. Additionally, switching participants from etanercept to the biosimilar during the subsequent treatment periods did not cause an increase in formation of antidrug antibodies.21
There are 2 upcoming studies involving biosimilars that are not yet recruiting patients. The first (NCT02925338) will analyze the characteristics of patients treated with Inflectra as well as their response to treatment. The second (NCT02762955) will be comparing the efficacy and safety of an adalimumab biosimilar (BCD-057, BIOCAD) to adalimumab.