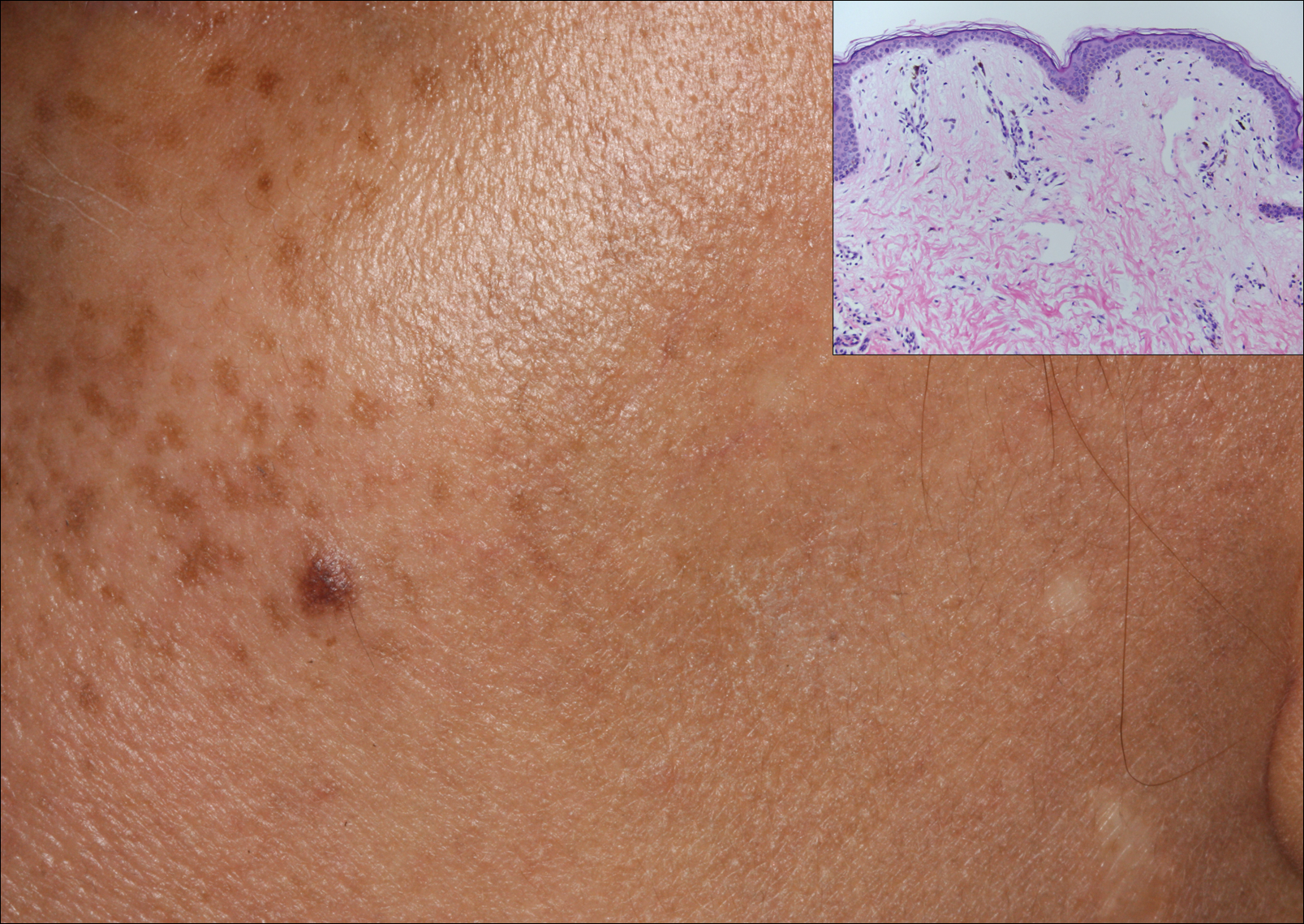
The Diagnosis: Erythema Dyschromicum Perstans
Erythema dyschromicum perstans (EDP), also referred to as ashy dermatosis, was first described by Ramirez1 in 1957 who labeled the patients los cenicientos (the ashen ones). It preferentially affects women in the second decade of life; however, patients of all ages can be affected, with reported cases occurring in children as young as 2 years of age.2 Most patients have Fitzpatrick skin type IV, mainly Amerindian, Hispanic South Asian, and Southwest Asian; however, there are cases reported worldwide.3 A genetic predisposition is proposed, as major histocompatibility complex genes associated with HLA-DR4⁎0407 are frequent in Mexican patients with ashy dermatosis and in the Amerindian population.4
The etiology of EDP is unknown. Various contributing factors have been reported including alimentary, occupational, and climatic factors,5,6 yet none have been conclusively demonstrated. High expression of CD36 (thrombospondin receptor not found in normal skin) in spinous and granular layers, CD94 (cytotoxic cell marker) in the basal cell layer and in the inflammatory dermal infiltrate,7 and focal keratinocytic expression of intercellular adhesion molecule I (CD54) in the active lesions of EDP, as well as the absence of these findings in normal skin, suggests an immunologic role in the development of the disease.8
Erythema dyschromicum perstans presents clinically with blue-gray hyperpigmented macules varying in size and shape and developing symmetrically in both sun-exposed and sun-protected areas of the face, neck, trunk, arms, and sometimes the dorsal hands (Figures 1 and 2). Notable sparing of the palms, soles, scalp, and mucous membranes occurs.
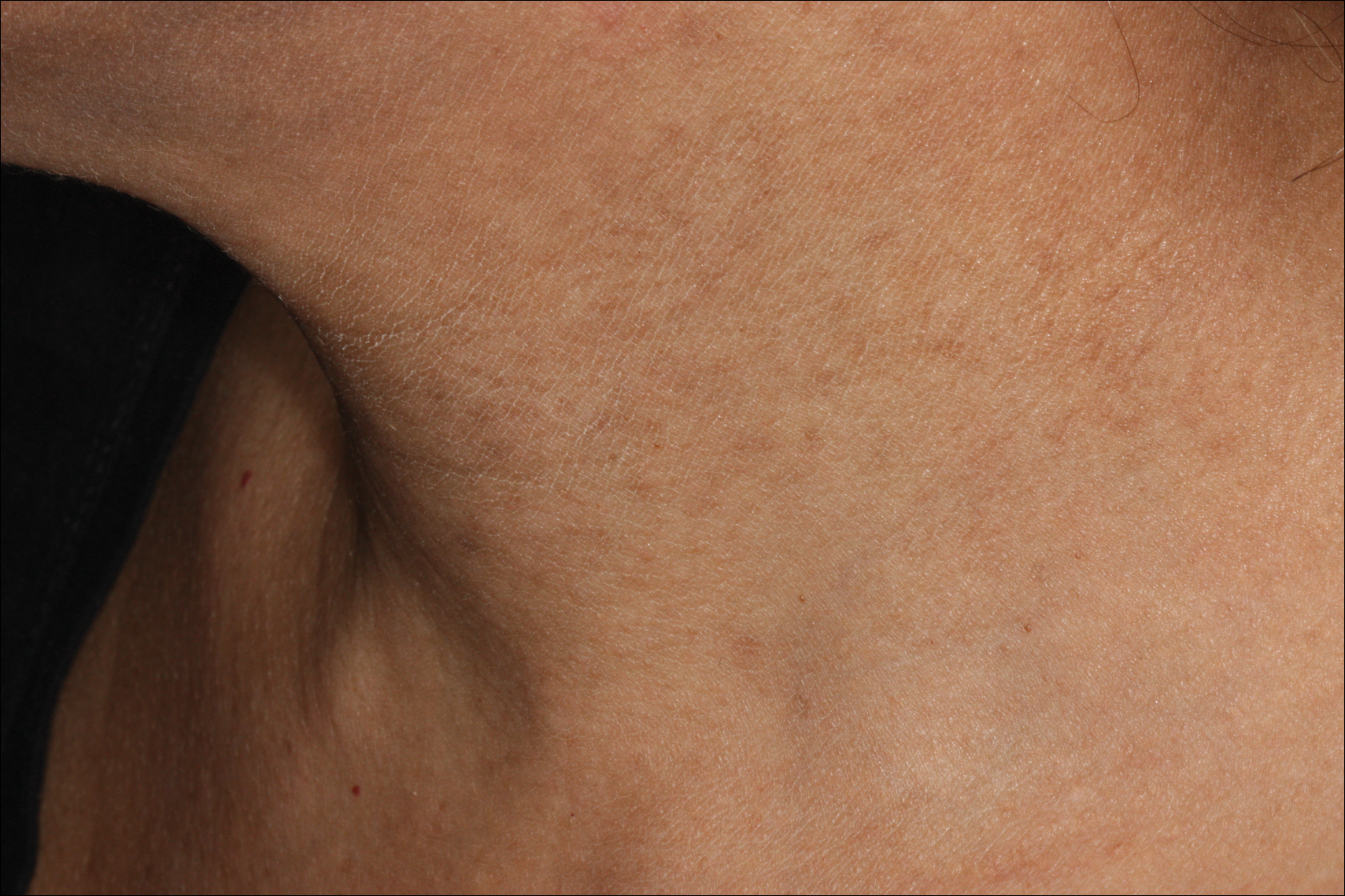
Figure 1. Blue-gray nonscaly macules and patches on the neck.
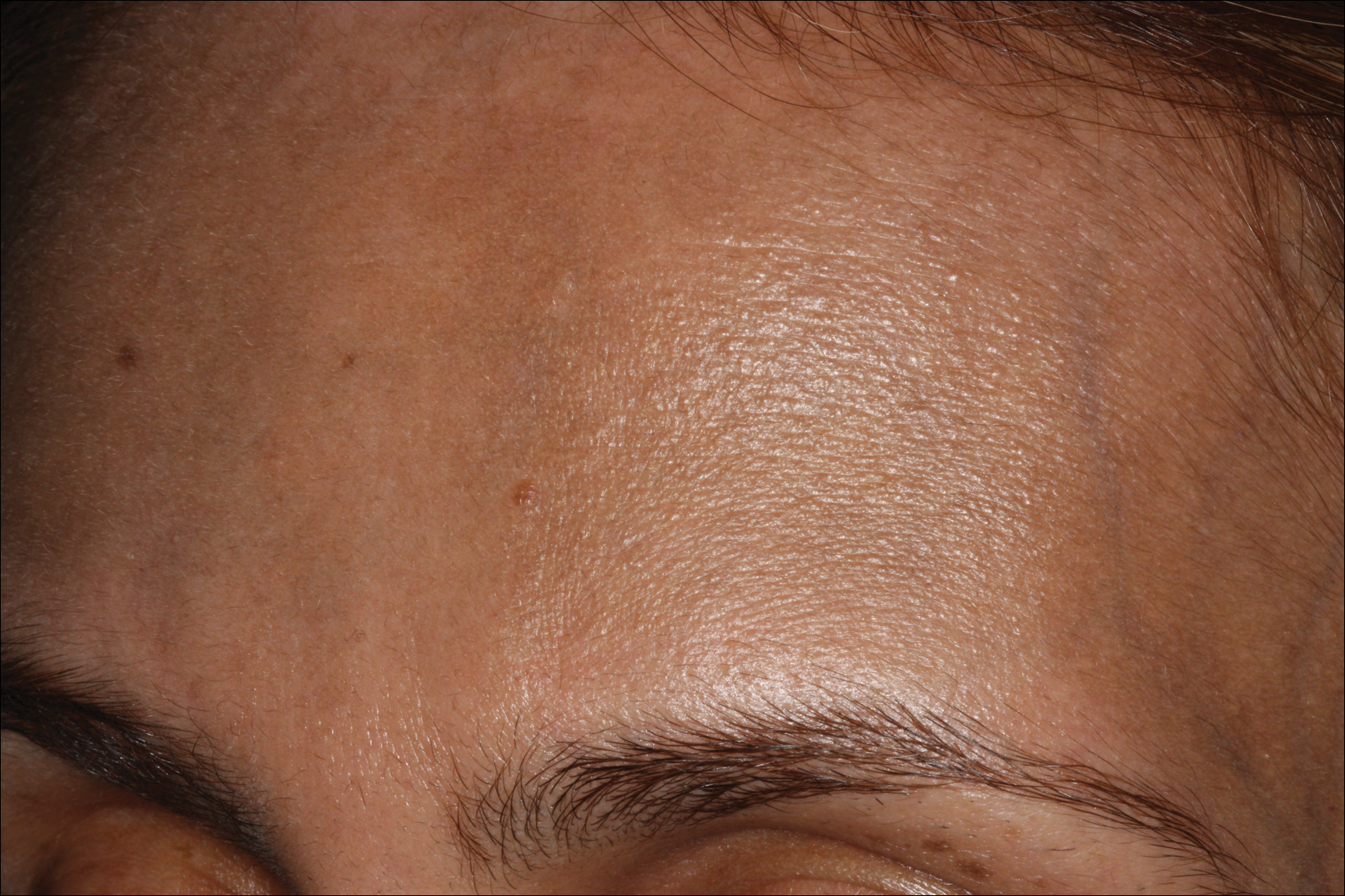
Figure 2. Bluish gray patches on the forehead.
Occasionally, in the early active stage of the disease, elevated erythematous borders are noted surrounding the hyperpigmented macules. Eventually a hypopigmented halo develops after a prolonged duration of disease.9 The eruption typically is chronic and asymptomatic, though some cases may be pruritic.10
Histopathologically, the early lesions of EDP with an erythematous active border reveal lichenoid dermatitis with basal vacuolar change and occasional Civatte bodies. A mild to moderate perivascular lymphohistiocytic infiltrate admixed with melanophages can be seen in the papillary dermis (Figure 3). In older lesions, the inflammatory infiltrate is sparse, and pigment incontinence consistent with postinflammatory pigmentation is prominent, though melanophages extending deep into the reticular dermis may aid in distinguishing EDP from other causes of postinflammatory pigment alteration.7,11
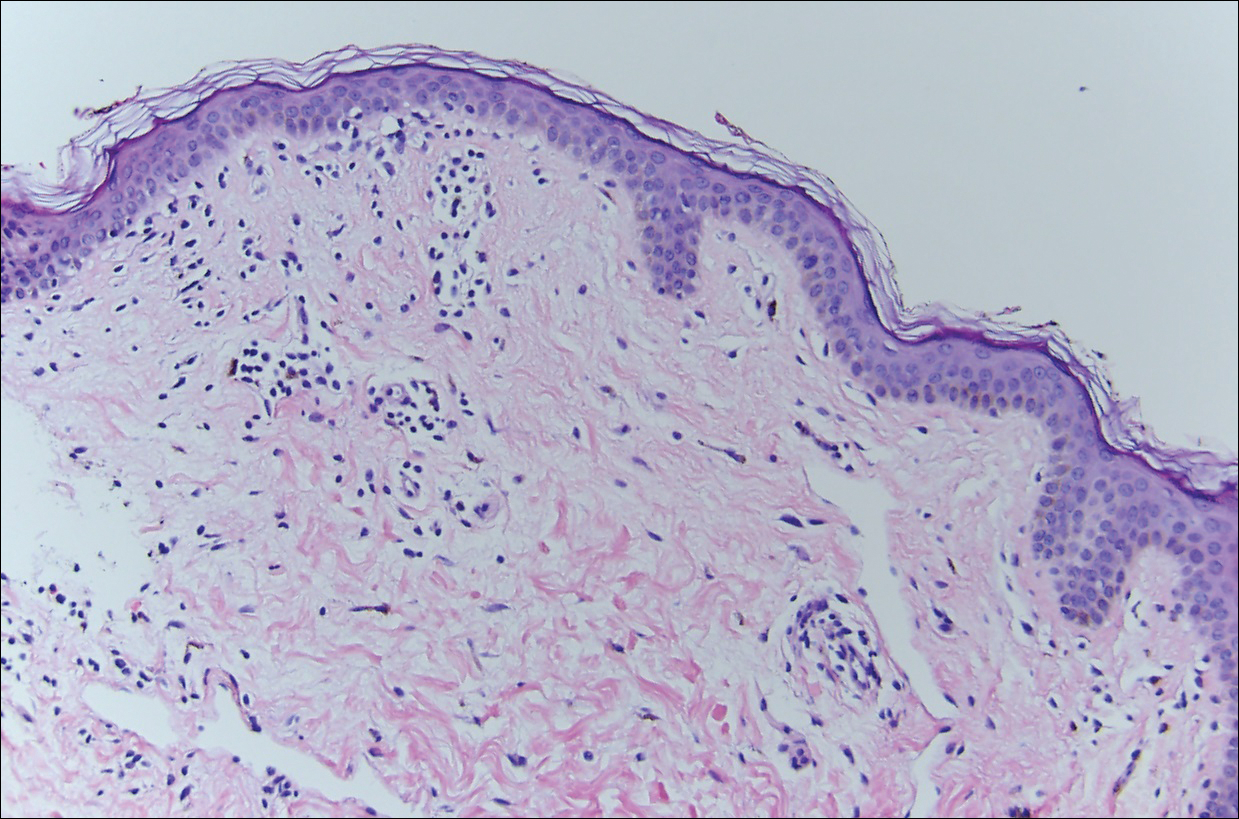
Figure 3. Subtle vacuolar interface dermatitis, perivascular lymphocytic infiltrate, and dermal melanophages (H&E, original magnification ×200).
Erythema dyschromicum perstans and lichen planus pigmentosus (LPP) may be indistinguishable histopathologically and may both be variants of lichen planus actinicus. Lichen planus pigmentosus often differs from EDP in that it presents with brown-black macules and patches often on the face and flexural areas. A subset of cases of LPP also may have mucous membrane involvement. The erythematous border that characterizes the active lesion of EDP is characteristically absent in LPP. In addition, pruritus often is reported with LPP. Direct immunofluorescence is not a beneficial tool in distinguishing the entities.12
Other differential diagnoses of predominantly facial hyperpigmentation include a lichenoid drug eruption; drug-induced hyperpigmentation (deposition disorder); postinflammatory hyperpigmentation following atopic dermatitis; contact dermatitis or photosensitivity reaction; early pinta; and cutaneous findings of systemic diseases manifesting with diffuse hyperpigmentation such as lupus erythematosus, dermatomyositis, hemochromatosis, and Addison disease. A detailed history including medication use, thorough clinical examination, and careful histopathologic evaluation will help distinguish these conditions.
Chrysiasis is a rare bluish to slate gray discoloration of the skin that predominantly occurs in sun-exposed areas. It is caused by chronic use of gold salts, which have been used to treat rheumatoid arthritis. UV light may contribute to induce the uptake of gold and subsequently stimulate tyrosinase activity.13 Histologic features of chrysiasis include dermal and perivascular gold deposition within the macrophages and endothelial cells as well as extracellular granules. It demonstrates an orange-red birefringence on fluorescent microscopy.14,15
Minocycline-induced hyperpigmentation is a well-recognized side effect of this drug. It is dose dependent and appears as a blue-black pigmentation that most frequently affects the shins, ankles, and arms.16 Three distinct types were documented: abnormal discoloration of the skin that has been linked to deposition of pigmented metabolites of minocycline producing blue-black pigmentation at the site of scarring or prior inflammation (type 1); blue-gray pigmentation affecting normal skin, mainly the legs (type 2); and elevated levels of melanin on the sun-exposed areas producing dirty skin syndrome (type 3).17,18
Topical and systemic corticosteroids, UV light therapy, oral dapsone, griseofulvin, retinoids, and clofazimine are reported as treatment options for ashy dermatosis, though results typically are disappointing.7