From the Department of Dermatology, Penn State Health Milton S. Hershey Medical Center.
The authors report no conflict of interest.
Correspondence: Taylor E. Gladys, BA, Penn State Health Milton S. Hershey Medical Center, 500 University Dr, HU14, Hershey, PA 17033 (tgladys@pennstatehealth.psu.edu).
A 71-year-old man presented with an eruption on the face, shoulders, upper back, and arms of 3 weeks’ duration. The lesions were asymptomatic, and he denied fever, chills, or weight loss. He had a history of type 2 diabetes mellitus, hypertension, and hypercholesterolemia. Physical examination revealed coarse facial features with purple-pink nodules on the face and trunk and ulcerated nodules on the upper extremities. Mucous membrane involvement was noted, and there was marked occipital and submandibular lymphadenopathy. A biopsy of an arm nodule revealed a superficial and deep dermal and periadnexal lymphocytic infiltrate of atypical CD3+ cells.
The Diagnosis: Primary Cutaneous γδ T-cell Lymphoma
Primary cutaneous γδ T-cell lymphoma (PCGDTL) is a distinct entity that can be confused with other types of cutaneous T-cell lymphomas. Often rapidly fatal, PCGDTL has a broad clinical spectrum that may include indolent variants—subcutaneous, epidermotropic, and dermal.1 Primary cutaneous γδ T-cell lymphoma represents less than 1% of all cutaneous T-cell lymphomas.2 Diagnosis and treatment remain challenging. Patients typically present with nodular lesions that progress to ulceration and necrosis. Early lesions can be confused with erythema nodosum, mycosis fungoides, or infection on clinical examination; biopsy establishes the diagnosis. Typical findings include a cytotoxic phenotype, variable epidermotropism, dermal and subcutaneous involvement, and loss of CD4 and often CD8 expression. Testing for Epstein-Barr virus expression yields negative results. The neoplastic lymphocytes in dermal and subcutaneous PCGDTL typically are T-cell intracellular antigen-1 (TIA-1) and granzyme positive.1
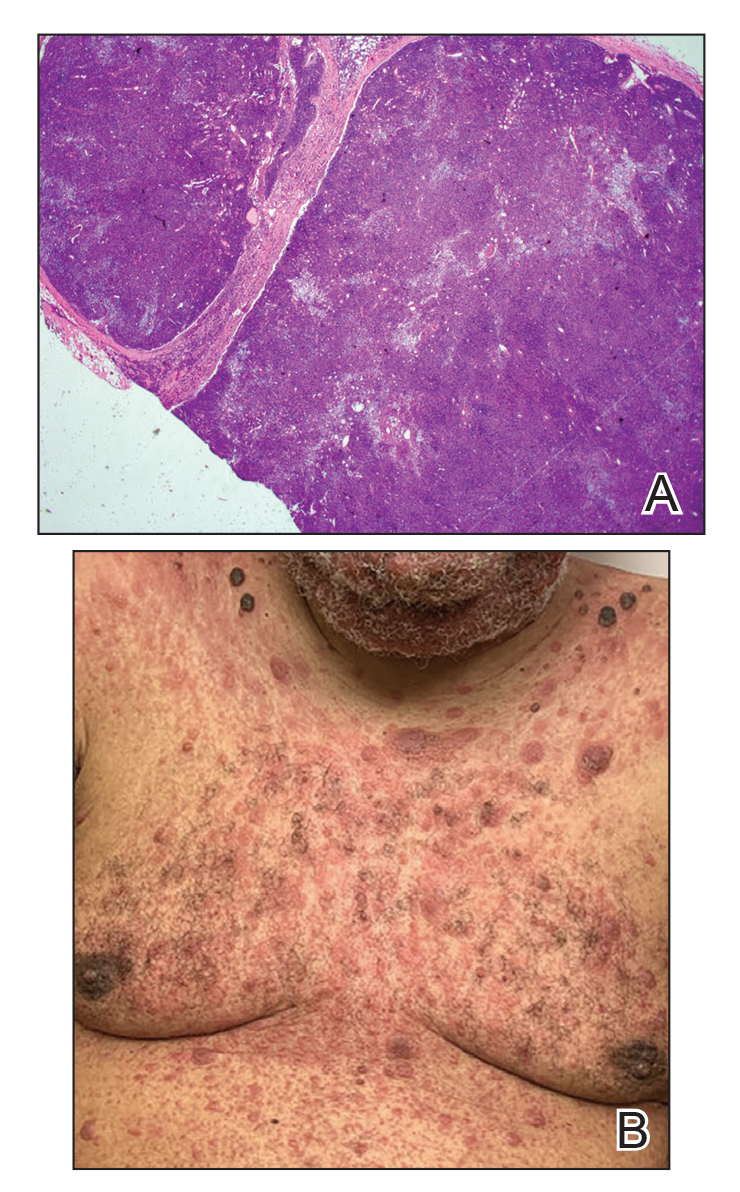
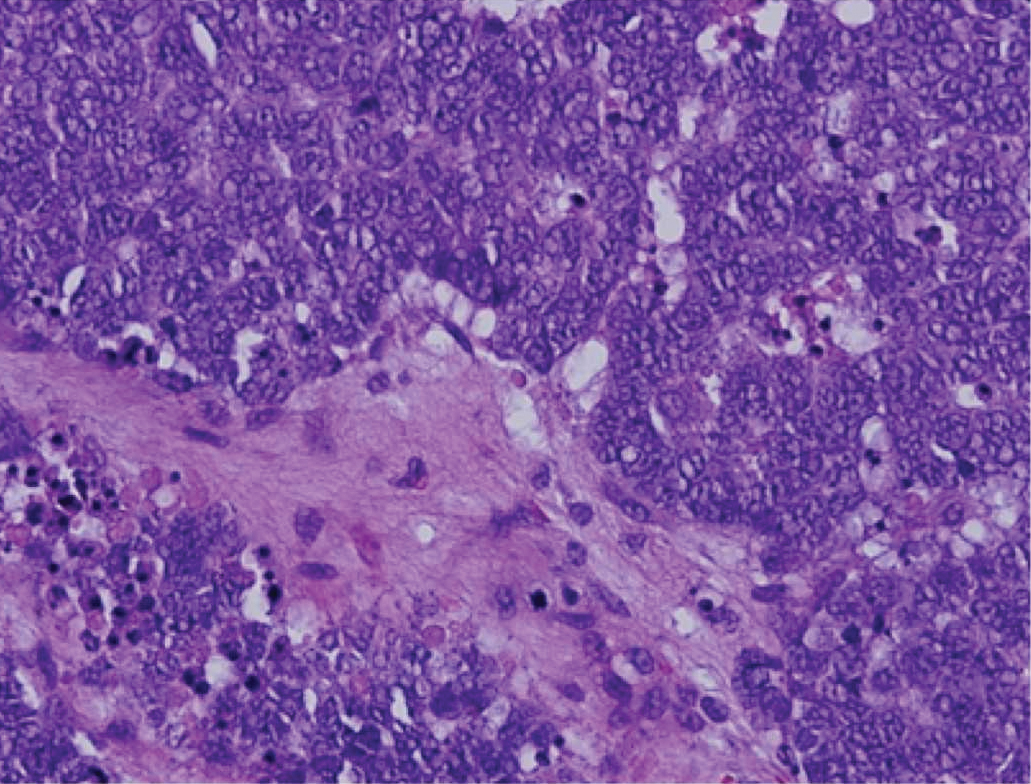
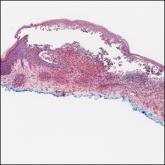
Immunohistochemistry failed to reveal CD8, CD56, granzyme, or T-cell intracellular antigen-1 staining of neoplastic cells in our patient but stained diffusely positive with CD3 and CD4. A CD20 stain decorated only a few dermal cells. The patient’s skin lesions continued to enlarge, and the massive lymphadenopathy made breathing difficult. Computed tomography revealed diffuse systemic involvement. An axillary lymph node biopsy revealed sinusoids with complete diffuse effacement of architecture as well as frequent mitotic figures and karyorrhectic debris (Figure 1A). Negative staining for T-cell receptor beta-F1 of the axillary lymph node biopsy and clonal rearrangement of the T-cell receptor gamma chain supported the diagnosis of PCGDTL. Nuclear staining for Epstein-Barr virus–encoded RNA was negative. Human T-cell leukemia virus type 1 antibodies and polymerase chain reaction also were negative. Flow cytometry demonstrated an atypical population of CD3+, CD4+, and CD7− γδ T lymphocytes, further supporting the diagnosis of lymphoma.
Figure 1. A, Axillary lymph node biopsy demonstrated visible sinusoids with complete diffuse effacement of architecture and frequent mitotic figures along with karyorrhectic debris (H&E, original magnification ×20). B, Leonine facies with erythematous papules and nodules distributed over the face, shoulders, and chest.
The median life expectancy for patients with dermal or subcutaneous PCGDTL is 10 to 15 months after diagnosis.3 The 5-year life expectancy for PCGDTL is approximately 11%.2 Limited treatment options contribute to the poor outcome. Chemotherapy regimens such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisolone) and EPOCH (etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) have yielded inconsistent results. Stem cell transplant has been tried in progressive disease and also has yielded mixed results.2 Brentuximab is indicated for individuals whose tumors express CD30.4 Associated hemophagic lymphohistiocytosis portends a poor prognosis.5
Despite treatment with etoposide, vincristine, doxorubicin, and high-dose oral steroids, our patient developed progressive difficulty breathing, stridor, kidney injury, and anemia. Our patient died less than 1 month after diagnosis—after only 1 round of chemotherapy—secondary to progressive disease and an uncontrollable gastrointestinal tract bleed. The leonine facies (Figure 1B) encountered in our patient can raise a differential diagnosis that includes infectious as well as neoplastic etiologies; however, most infectious etiologies associated with leonine facies manifest in a chronic fashion rather than with a sudden eruption, as noted in our patient.
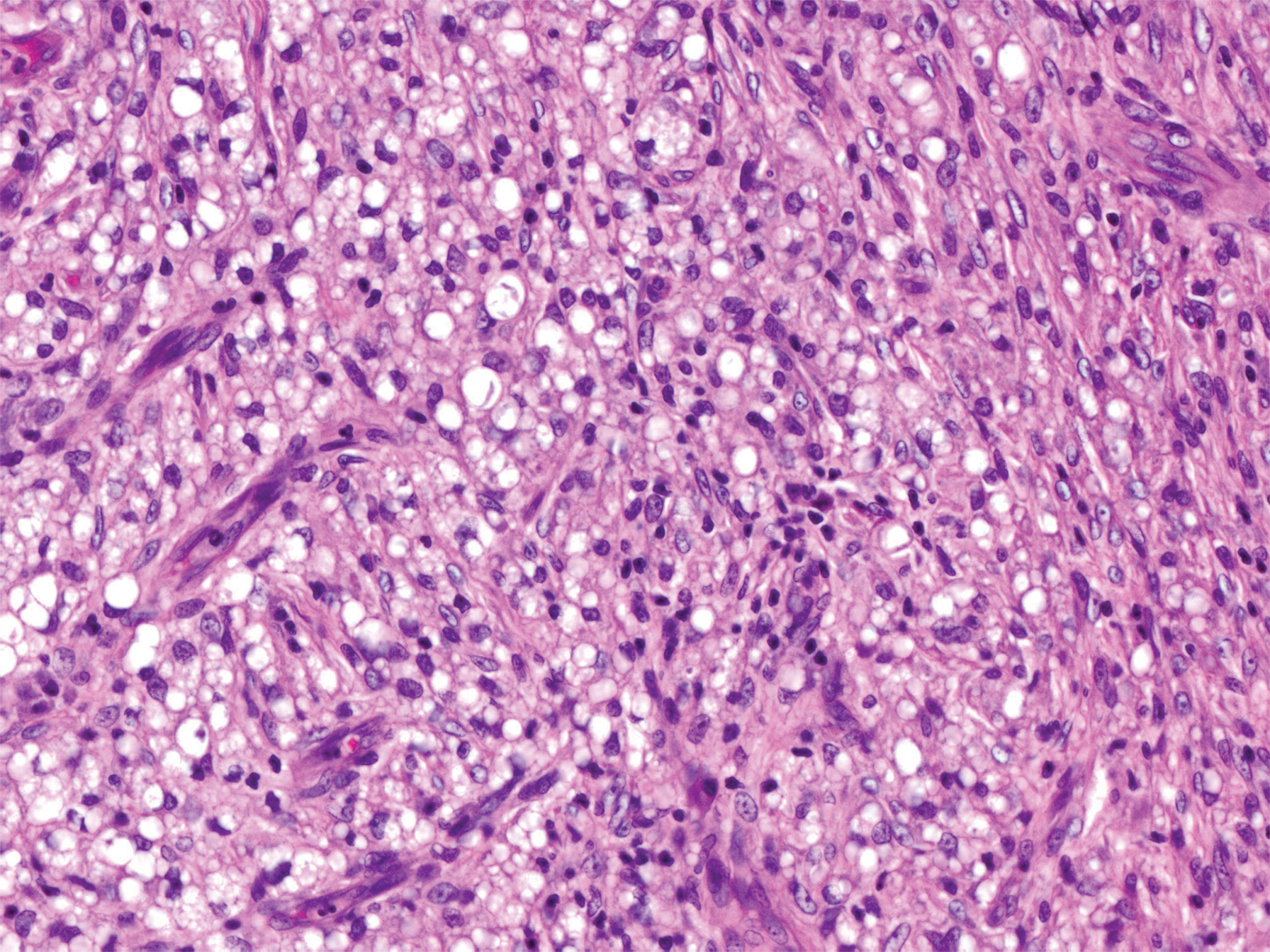
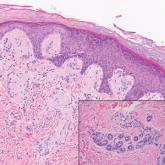
Leprosy is caused by Mycobacterium leprae, a grampositive bacillus. The condition manifests across a spectrum, with the poles being tuberculoid and lepromatous, and borderline variants in between.6-8 Lepromatous leprosy arises in individuals who are unable to mount cellular immunity against M leprae secondary to anergy.6 Lepromatous leprosy often presents with numerous papules and nodules. Aside from cutaneous manifestations, lepromatous leprosy has a predilection for peripheral nerves and specifically Schwann cells. Histologically, biopsy reveals a flat epidermis and a cell-free subepidermal grenz zone. Within the dermis, there is a diffuse histiocytic infiltrate that typically is not centered around nerves (Figure 2).6,7 Mycobacterium leprae can appear scattered throughout or clustered in globi. Mycobacterium leprae stains red with Ziehl-Neelsen or Wade-Fite stains.6,7 Immunohistochemistry reveals a CD4+ helper T cell (TH2) predominance, supported by the increased expression of type 2 reaction cytokines such as IL-4, IL-5, IL-10, and IL-13.8
Figure 2. Lepromatous leprosy. Dermis with a diffuse infiltrate of foamy histiocytes (H&E, original magnification ×400).
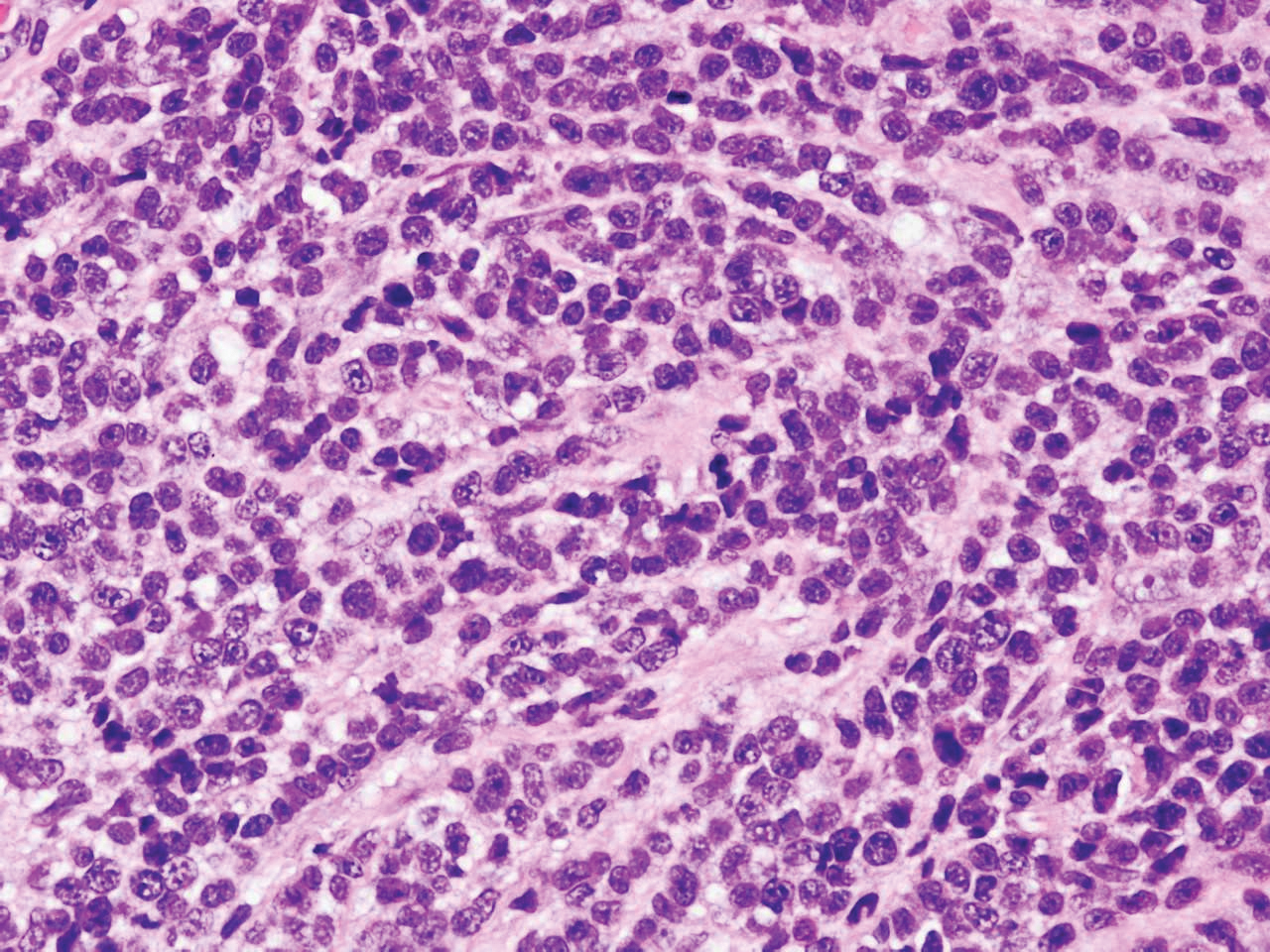
Diffuse large B-cell lymphoma (DLBCL) embodies 10% to 20% of all primary cutaneous lymphomas; it is more prevalent in older adults (age range, 70–82 years) and women. Clinically, DLBCL presents as either single or multiple rapidly progressing nodules or plaques, usually violaceous or blue-red in color.9,10 The most common area of presentation is on the legs, though it also can surface at other sites.9 On histology, DLBCL has clearly malignant features including frequent mitotic figures, large immunoblasts, and involvement throughout the dermis as well as perivascularly (Figure 3). Spindle-shaped cells and anaplastic features can be present. Immunohistochemically, DLBCL stains strongly positive for CD20 and B-cell lymphoma 2 (Bcl-2) along with other pan–B-cell markers.9-11 The aggressive leg type of DLBCL stains positively for multiple myeloma oncogene 1 (MUM-1).9,11
Figure 3. Diffuse large B-cell lymphoma. Widespread infiltration of immunoblasts with anaplastic features (H&E, original magnification ×400).
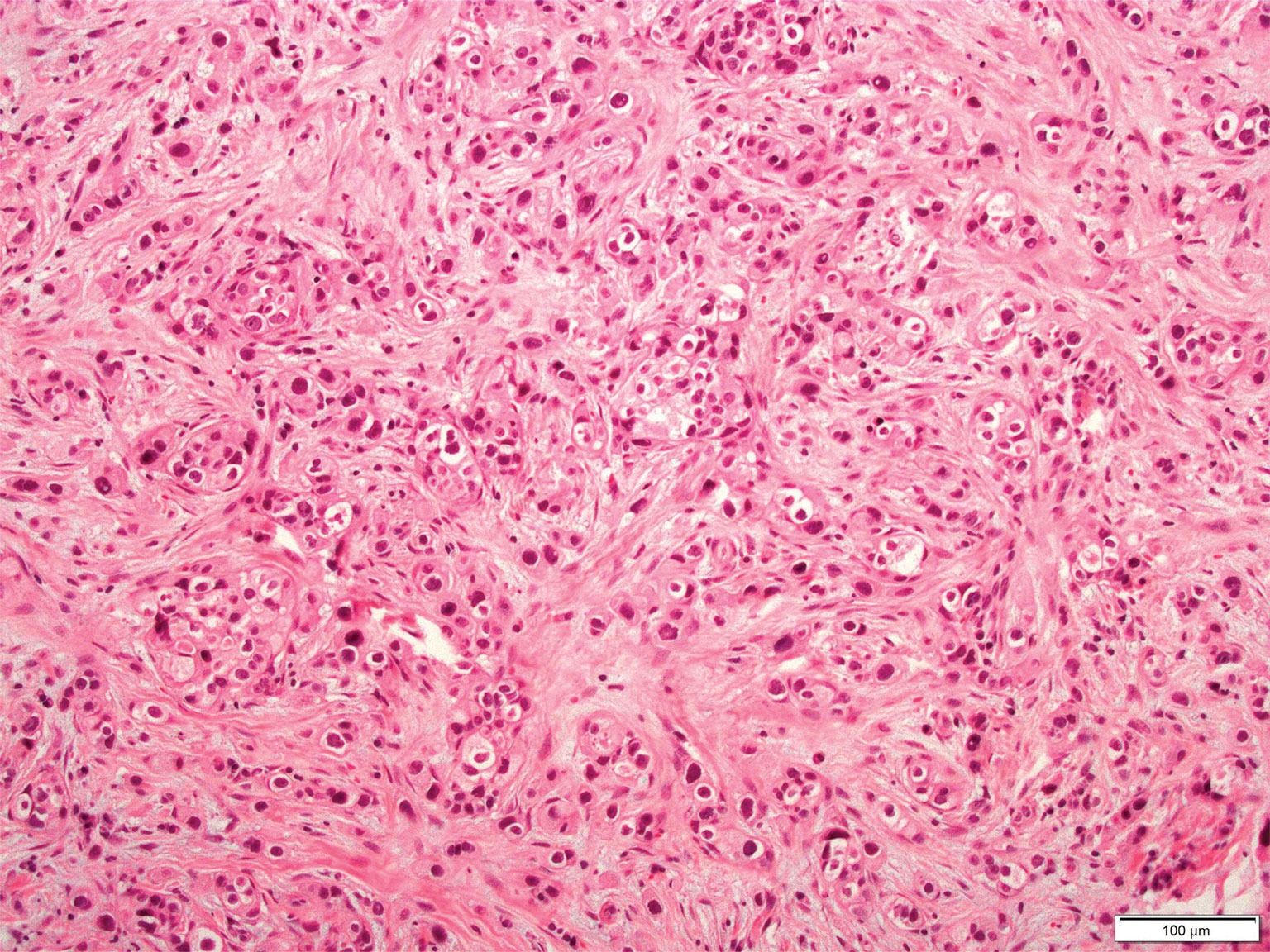
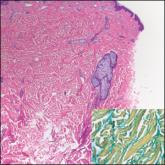
Cutaneous metastatic adenocarcinoma from internal malignancies occurs in approximately 5% of cancer patients with metastatic spread.12 Most of these cutaneous lesions develop in close proximity to the primary tumor such as on the trunk, head, or neck. All cutaneous metastases carry a poor prognosis. Clinical presentation can vary greatly, ranging from painless, firm, or elastic nodules to lesions that mimic inflammatory skin conditions such as erysipelas or scleroderma. The majority of these metastases develop as painless firm nodules that are flesh colored, pink, red-brown, or purple.12,13 The histopathology of metastatic adenocarcinoma demonstrates an infiltrative nodular appearance, though there rarely are well-circumscribed nodules found.13 The lesion originates in the dermis or subcutaneous tissue. It is a glandulartype lesion that may reflect the tissue of the primary tumor (Figure 4).12,14 Immunohistochemical stains likely will remain consistent with those of the primary tumor, which is not always the case.14
Figure 4. Metastatic adenocarcinoma. Dermis-based lesion with glandular features and loss of architecture (H&E, original magnification ×100).
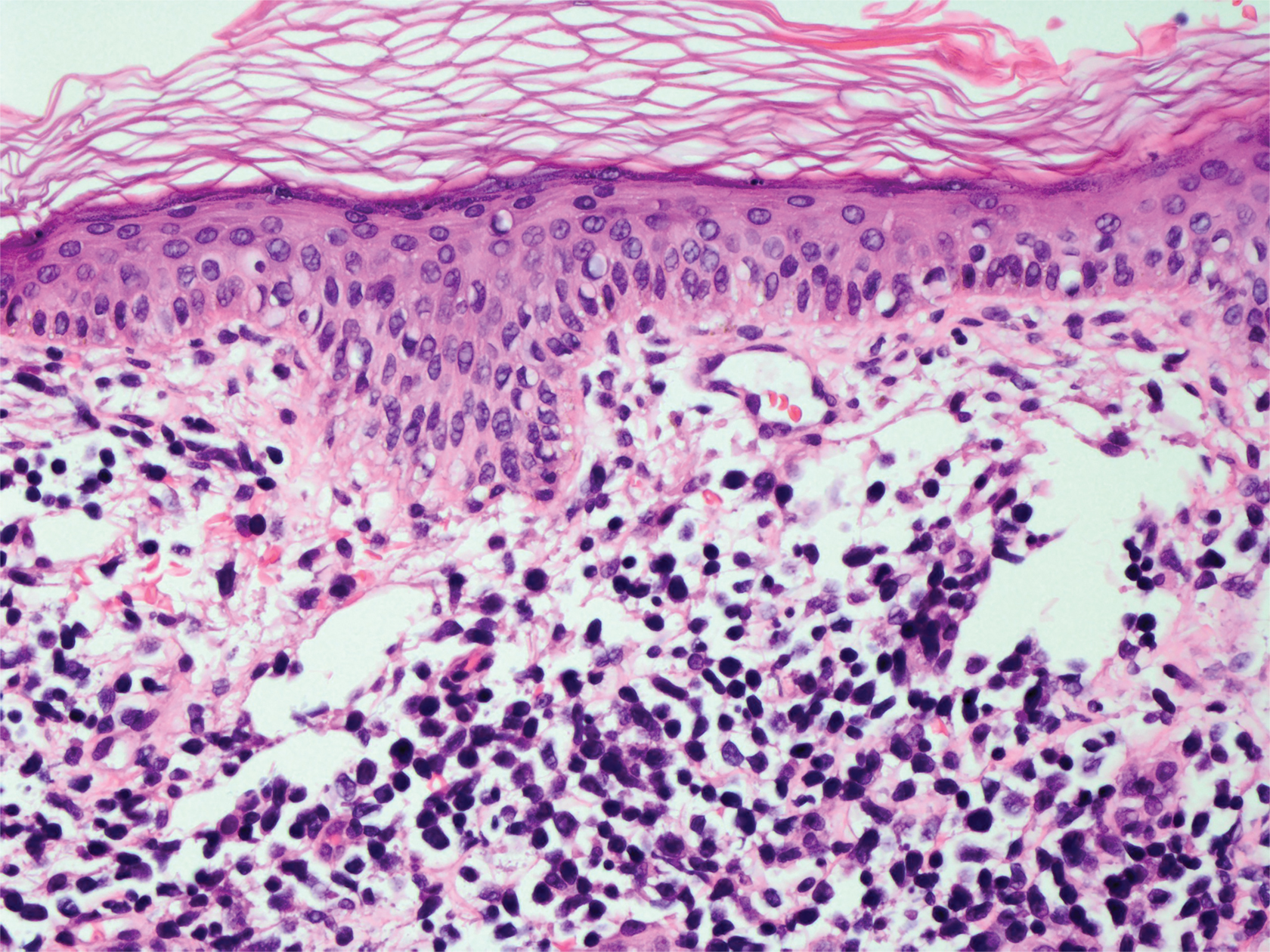
Merkel cell carcinoma (MCC) is an aggressive cutaneous malignancy of epithelial and neuroendocrine origin, first described as trabecular carcinoma due to the arrangement of tumor resembling cancellous bone.15,16 Merkel cells are mechanoreceptors found near nerve terminals.17 Approximately 80% of MCCs are associated with Merkel cell polyomavirus, which is a small, double-stranded DNA virus with an icosahedral capsid.17,18 Merkel cell polyomavirus–positive cases of MCC tend to have a better prognosis. In Merkel cell polyomavirus–negative MCC, there is an association with UV damage and increased chromosomal aberrations.18 Merkel cell carcinoma is known for its high rate of recurrence as well as local and distant metastasis. Nodal involvement is the most important prognostic indicator.15 Clinically, MCC is associated with the AEIOU mnemonic (asymptomatic, expanding rapidly, immunosuppression, older than 50 years, UV exposed/fair skin).15-17 Lesions appear as red-blue papules on sun-exposed skin and usually are smaller than 2 cm by their greatest dimension. On histopathology, MCC demonstrates small, round, blue cells arranged in sheets or nests originating in the dermis and occasionally can infiltrate the subcutis and lymphovascular surroundings (Figure 5).16-19 Cells have scant eosinophilic cytoplasm and may have fine granular chromatin. Numerous mitotic figures and apoptotic cells also are present. On immunohistochemistry, these cells will stain positive for cytokeratin AE1/AE3, anticytokeratin (CAM 5.2), CK20, and CD56. Due to their neuroendocrine derivation, they also are commonly synaptophysin, neuron-specific enolase, and chromogranin A positive. Notably, MCC will stain negative for leukocyte common antigen, CD20, CD3, CD34, and thyroid transcription factor 1 (TTF-1).16,17
Figure 5. Merkel cell carcinoma. Sheets of small, round, blue cells with granular chromatin, frequent mitotic figures, and apoptotic cells (H&E, original magnification ×400).
Primary cutaneous γδ T-cell lymphoma can be difficult to diagnose and requires urgent treatment. Clinicians and dermatopathologists need to work together to establish the diagnosis. There is a high mortality rate associated with PCGDTL, making prompt recognition and timely treatment critical. Acknowledgments—Thank you to our colleagues with the Penn State Health Hematology/Oncology Department (Hershey, Pennsylvania) for comanagement of this patient.
Acknowledgments Thank you to our colleagues with the Penn State Health Hematology/Oncology Department (Hershey, Pennsylvania) for comanagement of this patient.