To the Editor:
Relapsing polychondritis (RP) is a chronic, progressive, and episodic systemic inflammatory disease that primarily affects the cartilaginous structures of the ears and nose. Involvement of other proteoglycan-rich structures such as the joints, eyes, inner ears, blood vessels, heart, and kidneys also may be seen. Dermatologic manifestations occur in 35% to 50% of patients and may be the presenting sign in up to 12% of cases.1 The most commonly reported dermatologic findings include oral aphthosis, erythema nodosum, and purpura with vasculitic changes. Less commonly reported associations include Sweet syndrome, pyoderma gangrenosum, panniculitis, erythema elevatum diutinum, erythema annulare centrifugum, and erythema multiforme.1
A 43-year-old woman who was otherwise healthy developed new-onset tenderness and swelling of the left pinna while on vacation. She was treated with trimethoprim-sulfamethoxazole, clindamycin, and levofloxacin for presumed auricular cellulitis. The patient developed a fever; sore throat; and a progressive, pruritic, blistering rash on the face, torso, bilateral extremities, palms, and soles 1 day after completing the antibiotic course. After 5 days of unremitting symptoms despite oral, intramuscular, and topical steroids, the patient presented to the emergency department. Physical examination revealed diffuse, tender, erythematous to violaceous macules with varying degrees of coalescence on the chest, back, and extremities. Scattered flaccid bullae and erosions of the oral and genital mucosa also were seen. Laboratory analysis was notable only for a urinary tract infection with Klebsiella pneumoniae. A punch biopsy demonstrated full-thickness necrosis of the epidermis with subepidermal bullae and a mild to moderate lymphocytic infiltrate with rare eosinophils, consistent with a diagnosis of Stevens-Johnson syndrome (SJS). Because of the body surface area involved (20%) and the recent history of trimethoprim-sulfamethoxazole use, a diagnosis of SJS/toxic epidermal necrolysis (TEN) overlap syndrome was made. The patient was successfully treated with subcutaneous etanercept (50 mg), supportive care, and cephalexin for the urinary tract infection.
Approximately 5 weeks after discharge from the hospital, the patient was evaluated for new-onset pain and swelling of the right ear (Figure) in conjunction with recent tenderness and depression of the superior septal structure of the nose. A punch biopsy of the ear revealed mild perichondral inflammation without vasculitic changes and a superficial, deep perivascular, and periadnexal lymphoplasmacytic inflammatory infiltrate with scattered eosinophils. A diagnosis of RP was made, as the patient met Damiani and Levine’s2 criteria with bilateral auricular inflammation, ocular inflammation, and nasal chondritis.
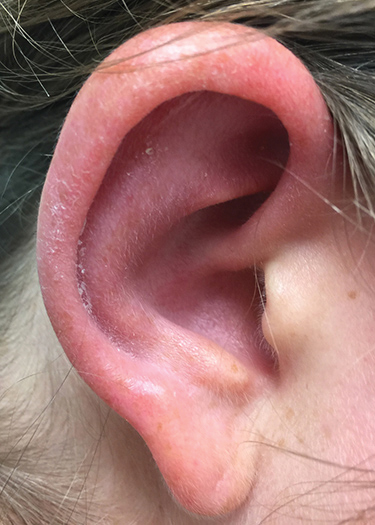
Erythema and tenderness of the right ear with characteristic sparing of the lobule.
Although the exact pathogenesis of RP remains unclear, there is strong evidence to suggest an underlying autoimmune etiology.3 Autoantibodies against type II collagen, in addition to other minor collagen and cartilage proteins, such as cartilage oligomeric matrix proteins and matrilin-1, are seen in a subset of patients. Titers have been reported to correlate with disease activity.3,4 Direct immunofluorescence also has demonstrated plentiful CD4+ T cells, as well as IgM, IgA, IgG, and C3 deposits in the inflamed cartilage of patients with RP.3 Additionally, approximately 30% of patients with RP will have another autoimmune disease, and more than 50% of patients with RP carry the HLA-DR4 antigen.3 Alternatively, SJS and TEN are not reported in association with autoimmune diseases and are believed to be CD8+ T-cell driven. Some HLA-B subtypes have been found in strong association with SJS and TEN, suggesting the role of a potential genetic susceptibility.5
We report a unique case of SJS/TEN overlap syndrome occurring in a patient with RP.1 Although the association may be coincidental, it is well known that patients with lupus erythematosus are predisposed to the development of SJS and TEN. Therefore, a shared underlying genetic predisposition or immune system hyperactivity secondary to active RP is a possible explanation for our patient’s unique presentation.