From the University of California Los Angles, David Geffen School of Medicine. Dr. Hiscox is from the Department of Medicine, and Drs. Hu and Young are from the Division of Dermatology.
The authors report no conflict of interest.
Correspondence: Lorraine C. Young, MD, Department of Medicine, University of California at Los Angeles (UCLA) David Geffen School of Medicine, 200 UCLA Medical Plaza, Ste 450, Los Angeles, CA 90095 (lcyoung@mednet.ucla.edu).
This report represents the second documented case of Morfan syndrome. Because of its extreme rarity, there is no specific diagnostic algorithm for Morfan syndrome.
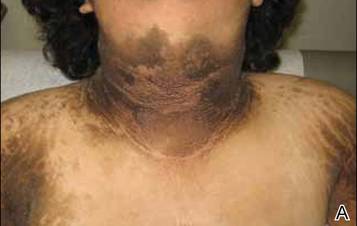
A 17-year-old adolescent girl presented to our clinic for evaluation of diffuse acanthosis nigricans (AN) that had been present since 7 years of age. The patient had a history of hypothyroidism, insulin resistance, ovarian cysts, and developmental delay. On examination, she presented with thick and verrucous plaques of AN involving the neck, abdomen, trunk, arms, and legs (Figure). The intertriginous areas were affected the most. The examination also was notable for dysmorphic facies and an endomorphic body habitus. Both parents denied similar health problems in their family and were normal in appearance. Although the patient was receiving metformin treatment for insulin resistance, she had not undergone any prior workup to identify a unifying syndromic cause for her physical and biochemical findings. A review of the literature showed a 1993 case report of a 5-year-old boy with mental retardation, body overgrowth, remarkable facies, and AN, which was termed Morfan syndrome.1 Because of the similarity in features, we believe that our patient’s presentation fits this syndrome. This report represents the second documented case of Morfan syndrome according to a PubMed search of articles indexed for MEDLINE using the search term Morfan. Additional searches using the terms acanthosis nigricans and syndrome also failed to identify any reports describing patients with a similar constellation of findings.
Thick and verrucous brown plaques involving the neck and upper arms (A) as well as the trunk (B).
First described by Santi Unna and Monatsh Pollitzer in 1890, AN is a common dermatosis that is characterized by thick, hyperpigmented, and verrucous plaques.2,3 Although most common in symmetric distribution on flexural and intertriginous areas, AN also may involve mucosal surfaces.4 Acanthosis nigricans is associated with multiple etiologic factors, including direct autosomal transmission, genetic abnormalities, medications, malignancy, and endocrine imbalance.2 However, the diffuse generalized form of AN is almost always found in the context of malignancy or genetic syndromes.5 Historically, most attention on AN has focused on its eruptive form (so-called malignant AN), which is usually associated with internal malignancy but also may result from benign pituitary adenomas.6 Although the precise mechanism for paraneoplastic AN is still a matter of debate, it is likely the result of overactive growth factors, such as transforming growth factor α, epidermal growth factor, and α melanocyte-stimulating hormone.7 Generalized AN also has been associated with genetic abnormalities. Multiple genetic mutations have been associated with AN, including the genes coding for the insulin receptor, fibroblast growth receptors 2 and 3, lamin A/C, and seipin.8-12 Acanthosis nigricans has been described as a feature in other genetic syndromes but may represent an incidental finding.4 In 1976, Kahn et al13 linked AN with insulin resistance, which subsequently shifted the focus on AN’s link with obesity and as a precursor of type 2 diabetes mellitus.6,14 In these cases, it is hypothesized that excess insulin activates the insulinlike growth factor 1 receptor to stimulate keratinocyte and dermal fibroblast proliferation. It is likely that hyperinsulinemia also induces cellular proliferation indirectly through other pathways.15,16 Compared with paraneoplastic and syndromic causes, AN secondary to hyperinsulinemia and obesity does not tend to be generalized but instead is proportionate to the degree of metabolic derangement.17
Because our patient’s AN presentation was diffuse, long-standing, and occurred in the context of morphologic abnormalities, it was consistent with a syndrome. Based on the constellation of findings, we believe our patient fit the rare markers indicating Morfan syndrome. Because of its extreme rarity, there is no specific diagnostic algorithm for Morfan syndrome. As additional cases are reported, we hope that further biochemical, physical, and genetic studies may be pursued to better identify the syndrome and elucidate its pathogenesis.