The Diagnosis: Tuberous Sclerosis Complex
Tuberous sclerosis complex (TSC) is an autosomal-dominant neurocutaneous syndrome characterized by multiple hamartomas distributed in multiple organs of the body, most commonly the skin, brain, eyes, heart, kidneys, liver, and lungs. In 1908, Vogt1 elucidated the classic diagnostic triad of seizures, mental retardation, and facial angiofibromas (formerly termed adenoma sebaceum). However, the full triad is evident in only 29% of cases; 6% of TSC patients have none of these 3 findings.2 The disease is caused by alterations in 2 TSC genes, TSC1 and TSC2, on chromosomes 9 and 16, respectively. Both are tumor suppressor genes and mutations in either can be responsible for all the complications seen in the disease.3 The gene products of the 2 genes, hamartin and tuberin, appear to act together in the regulation of cell growth by inhibiting a substance known as the mechanistic target of rapamycin.4,5 Up to two-thirds of cases may occur from spontaneous mutations.2,6 The disease is extremely variable in its manifestations, particularly the skin manifestations. This discussion will review the many cutaneous manifestations of TSC, most of them documented in our patient, and their frequency of occurrence.
Early recognition of TSC is vital because prompt implementation of the recommended diagnostic evaluation (eg, neuroimaging studies, electroencephalogram, electrocardiography, renal ultrasonography, chest computed tomography) may prevent serious clinical consequences.7 Neurologic manifestations are the leading cause of morbidity and mortality in patients with TSC.2,8,9 Brain hamartomas in the form of cortical tubers, subependymal nodules, and subependymal giant cell astrocytomas often are responsible for intractable seizures, most commonly as infantile spasms. Approximately 90% to 96% of TSC patients have seizures.6 Renal manifestations are strongly associated with TSC. Angiomyolipoma is the most common renal lesion found in TSC patients.4,9,10 Up to 80% of patients have renal angiomyolipomas. Their incidence increases with age.4,11,12 Tuberous sclerosis complex is known as a cause of epilepsy and mental retardation, but renal disease is a major cause of morbidity.13 Cardiovascular manifestations often are the earliest diagnostic findings in patients with TSC. Rhabdomyoma is the most common primary cardiac tumor in infants and children.11,13,14 The most common ocular findings in TSC are retinal hamartomas, appearing in 40% to 50% of patients.15
Cutaneous features frequently occur and are the most clinically apparent findings of TSC; if overlooked, diagnosis could be delayed, which ultimately increases mortality. The diagnosis of TSC continues to be based primarily on clinical grounds because most patients older than 5 years demonstrate multiple skin lesions.6 In 1992, specific diagnostic clinical criteria were created for TSC, which stratified the clinical features of TSC into 3 tiers—primary, secondary, and tertiary—based on their specificity.16 In subsequent years, some of the classic clinical signs once regarded as pathognomonic for TSC, such as single ungual fibroma or multiple renal angiomyolipomas, were questioned. New modifications to the original diagnostic criteria were necessary given the advances in both TSC clinical information and molecular genetics. In the summer of 1998, a consensus conference held in Annapolis, Maryland, was assembled by the Tuberous Sclerosis Alliance for the purpose of reevaluating and updating the clinical diagnostic criteria.7 The revised criteria were simplified into 2 main categories—major and minor features—based on the diagnostic importance and degree of specificity for TSC of each clinical and radiographic feature (Table 1). There are a number of considerations that merit close attention when applying these clinical criteria. Despite the large number of clinical features delineated, the diagnosis of TSC may be challenging early in life, particularly in patients younger than 2 years. The value of the clinical criteria for early diagnosis and prompt management of TSC is limited by the fact that many stigmata become apparent only in late childhood or adulthood. The lack of pathognomonic signs of TSC can add to the challenge of diagnosing subtle or atypical cases.6

A careful skin examination of individuals at risk for TSC is the best and the easiest method of establishing the diagnosis in most cases. According to Gomez,17 96% of patients with TSC have one or more main skin lesions of the disease, including facial angiofibromas, ungual fibromas, shagreen patches, or hypomelanotic macules. However, the diagnosis is more difficult in small children because many skin lesions become obvious with age; in fact, the full dermatological spectrum may never develop.17
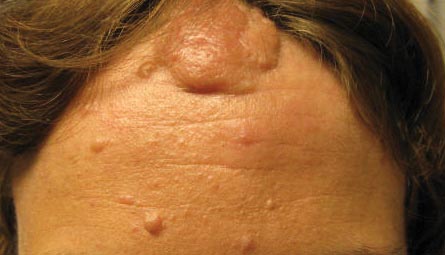
Facial angiofibromas, also known as sebaceous adenomas, are the most visible and unsightly cutaneous manifestations of TSC, often resulting in stigmatization for both the affected individuals and their family members. These facial lesions often do not appear until late childhood or adulthood. In this case, the initial diagnosis often is made by the dermatologist who appreciates the significance of the white macules in a neonate. However, these hypomelanotic macules cannot be detected by the unaided eye, thus a physician would be wise to examine an infant with infantile spasms or mental retardation with a Wood lamp, which accentuates the lesions.2