Early and aggressive therapy may prevent or stabilize organ damage and disease-related complications in patients with juvenile dermatomyositis, according to new consensus-based recommendations for the management of the disorder.
Overall, the recommendations – a project of the Single Hub and Access Point for Pediatric Rheumatology in Europe (SHARE) initiative – include 7 overarching principles, 33 recommendations on diagnosis, and 19 recommendations on therapy that were accepted with greater than 80% agreement among experts. The recommendations, which address the assessment of skin, muscle, and major organ involvement and treatment suggestions at disease onset and in refractory cases, fill a void in the area of evidence-based guidelines for this rare disease within the group of pediatric rheumatic diseases, according to Felicitas Bellutti Enders, MD, of University Medical Center Utrecht (the Netherlands) and her colleagues.
 Elizabeth M. Dugan, Adam M. Huber, Frederick W. Miller, Lisa G. Rider / CC-BY-SA-3.0
Elizabeth M. Dugan, Adam M. Huber, Frederick W. Miller, Lisa G. Rider / CC-BY-SA-3.0
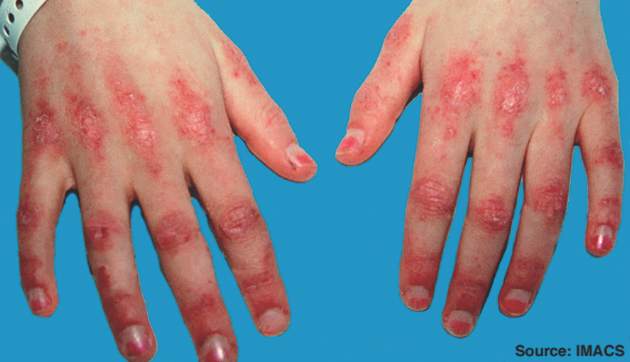
A patient with juvenile dermatomyositis shows Gottron's papules, erythematous to violaceous raised papules overlying the metacarpal and interphalangeal joints.
“Clear recommendations can help clinicians in the care of patients with JDM [juvenile dermatomyositis] as no international consensus regarding diagnosis and treatment is currently available and management therefore varies,” they wrote (Ann Rheum Dis. 2016 Aug 11. doi: 10.1136/annrheumdis-2016-209247).
The consensus committee, including 19 experienced pediatric rheumatologists and 2 experts in pediatric exercise physiology and physical therapy, developed recommendations based on a validated systematic literature review. The recommendations were evaluated by an online survey and then discussed at two subsequent consensus meetings.
The overarching principles accepted by more than 80% of respondents (100% in all but number 4 below), hold that:
1) All children with suspected idiopathic inflammatory myopathies should be referred to a specialized center.
2) High-risk patients need immediate/urgent referral to a specialized center.
3) Patient-/parent-reported outcomes measures are helpful when assessing disease activity and should be used at diagnosis and during disease monitoring.
4) Validated tools such as the Childhood Health Assessment Questionnaire, patient/parent visual analog scale and Juvenile Dermatomyositis Multi-dimensional Assessment Report should be used to measure health status.
5) All children with JDM should have disease activity (muscle, skin, major organ) assessed regularly in a standardized way, using tools such as the Disease Activity Score.
6) All children with JDM should have disease damage assessed at least yearly using a standardized disease damage measure, such as the Myositis Damage index.
7) All patients with JDM should have the opportunity to be registered within a research registry/repository such as the Euromyositis registry.
The consensus process also yielded both general and specific recommendations for diagnosis and management – also with 100% agreement in almost all cases – addressing investigations to consider in all those with a JDM diagnosis (muscle enzymes, full blood count, renal function and liver function tests, and infection screen – to name a few), ways that MRI can be used (both for diagnosis and disease activity monitoring), the use of muscle biopsy (use in all cases involving atypical presentation), and assessment of calcinosis and skin, lung, and cardiac involvement.
Treatment recommendations address sun protection (encourage routine use of sunblock), exercise (should be safe and appropriate and monitored by a physiotherapist), corticosteroid use (administer systematically either orally or intravenously in moderate to severe JDM and wean as the patient shows clinical improvement), use of intravenous immunoglobulin (a useful adjunct for resistant disease, especially when skin features are prominent), and the use of anti–tumor necrosis factor therapies (consider in refractory disease; infliximab or adalimumab are favored over etanercept), as well as other treatments.
The recommendations state that while there is no high-level evidence of when to stop therapy, consideration may be given to withdrawing treatment if a patient has been off steroids and in remission on methotrexate or an alternative disease-modifying antirheumatic drug [DMARD] for at least 1 year.
The management of JDM is complex and warrants a multidisciplinary approach, involving physiotherapists, specialist nurses, pediatric rheumatologists, and other specialists as needed, the authors wrote, noting that the mainstay of therapy is high-dose corticosteroids initially in combination with DMARDS.
However, the evidence base for treatment is limited and “often confined to small case-controlled studies, with the exception of two randomized controlled trials,” they said.
Similarly, there is no high-level evidence regarding when to stop immunosuppressive therapy, but the expert group suggested considering the withdrawal of methotrexate or alternative DMARD once the patient is in remission and off steroids for a minimum of 1 year.
The authors concluded that “this SHARE initiative is based on expert opinion informed by the best available evidence and provides recommendations ... with a view to improving the outcome for patients with JDM in Europe.