Comment
Clinical Presentation
Lichen planus pemphigoides is a rare autoimmune subepidermal blistering disease with few cases reported in the literature. It is considered a clinical variation of bullous pemphigoid (BP) or a coexistence of LP and BP.1,2 It is characterized by bullous lesions developing on LP papules as well as on clinically uninvolved areas of the skin. It has been reported that LPP is provoked by several medications including cinnarizine, captopril, ramipril, simvastatin, psoralen plus UVA, and antituberculous medications (eg, isoniazid, rifampin, ethambutol, pyrazinamide).1 Risedronate or atenolol have not been reported to cause LPP, LP, or BP; however, according to Litt,3 a lichenoid drug eruption has been associated with atenolol. Furthermore, some cases of LPP demonstrate overlapping characteristics with paraneoplastic pemphigus and have been associated with internal malignancy. Hamada et al4 described a case of LPP coupled with colon adenocarcinoma and numerous keratoacanthomas. The earliest depiction of the coexistence of a case of mainstream LP complicated by an extensive bullous eruption was by Kaposi5 in 1892. He coined the term lichen ruber pemphigoides.5
Compared to BP, LPP is believed to affect a younger age group and have a less serious clinical course. The mean age of onset of LPP is in the third to fourth decades of life, while BP typically presents in the sixth decade. When comparing the location of bullae in LPP versus BP, the lesions of LPP tend to occur on the limbs, while BP tends to occur on the trunk.6
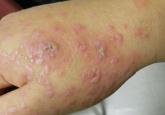
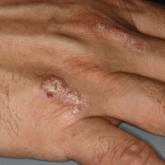
Clinically, LPP is distinguished by the existence of bullous lesions developing atop of the lesions of LP as well as on normal skin, with the latter being more commonplace. A classic example of LPP is characterized by an initial episode of traditional LP lesions often having severe pruritus, with or without patches of erythema, with the sudden eruption of tense bullae. These bullae commonly appear on the extremities and can appear over the normal skin, erythematous patches, or preexisting papules.7 In the atypical clinical presentations of this dubious skin condition, the bullae may only be seen on the lesions of LP.8 There also could be a lichenoid erythrodermic manifestation of a bullous eruption.9
Oral lesions of LPP have been described but had not been studied immunopathologically until Allen et al10 portrayed a 59-year-old man with cutaneous and oral lesions of LPP. They performed biopsies on the oral lesions and examined them by routine light microscopy and immunofluorescent techniques. The fine keratotic striae on the anterior buccal mucosal lesions were clinically consistent with oral LP. Perilesional tissue in conjunction with ulceration of the posterior buccal mucosa demonstrated histologic and immunopathologic alterations consistent with BP.10
Histopathology
Histopathologically, the lesions of LP show a bandlike lymphohistiocytic infiltrate, colloid bodies in the dermis, irregular acanthosis with saw-toothed rete ridges, orthokeratosis, wedge-shaped hypergranulosis, and liquefaction degeneration of the basal layer. Direct immunofluorescence shows mainly IgM and C3 deposited on colloid bodies, fibrin, and fibrinogen.11 The histopathology of the bullous lesion of LPP depicts a subepidermal bulla with variable diffuse or sparse lymphohistiocytic infiltrate and frequent eosinophils with or without neutrophils in the upper dermis. The existence of C3 alone or with IgG along the dermoepidermal junction gives confirmation on DIF.7
Autoantibodies
The expression of IgG autoantibodies directed against the basement membrane zone distinguishes LPP from bullous LP.2 IgG autoantibodies to either one or both the 230-kDa and 180-kDa BP (type XVII collagen) antigens has been demonstrated with LPP.4,12-14 Hamada et al4 described a histologic pattern more consistent with paraneoplastic pemphigus. It has been suggested that injury to the basal cells in LP or damage due to other courses of therapy such as psoralen plus UVA unveil suppressed antigenic determinants or produce new antigens, leading to antibody development and production of BP.12,15
Zillikens et al2 performed a study to identify the target antigen of LPP autoantibodies. They used sera from patients with LPP (n=4) and stained the epidermal side of salt-split human skin in a configuration identical to BP sera. In BP, the autoimmune response is directed against BP180, a hemidesmosomal transmembrane collagenous glycoprotein. They demonstrated that sera from BP patients largely reacted with a set of 4 epitopes (MCW-0 through MCW-3) grouped within a 45 amino acid stretch of the major noncollagenous extracellular domain (NC16A) of BP180. By immunoblotting and enzyme-linked immunosorbent assay, LPP sera also were compellingly reactive with recombinant BP180 NC16A. Lichen planus pemphigoides epitopes were additionally mapped using a series of overlapping recombinant segments of the NC16A domain. The authors demonstrated that all LPP sera reacted with amino acids 46 through 59 of domain NC16A, a protein portion that was previously shown to be unreactive with BP sera. In addition, they showed that 2 LPP sera reacted with the immunodominant antigenic region related to BP. Furthermore, they identified a unique epitope within the BP180 NC16A domain—MCW-4—which was distinctively recognized by sera from patients with LPP.2
Pathogenesis
The pathogenesis of both LP and BP has been linked to multiple cytokines that induce apoptosis in basal keratinocytes. Implicated cytokines include IFN-γ, tumor necrosis factor α (TNF-α), IL-1, IL-6, and IL-8, as well as other apoptosis-related molecules, such as Fas/Apo-1 and Bcl-2 in LP.16-18 Soluble E-selectin, vascular endothelial growth factor, IL-1β, IL-8, IL-5, transforming growth factor β1, and TNF-α were found to be elevated in either blister fluid or sera of BP patients.15-17
Management
Lichen planus pemphigoides usually responds well to traditional therapies, with systemic steroids being the most efficacious treatment of extensive disease.12,13 Other options include tetracycline and nicotinamide, isotretinoin, dapsone, and immunosuppressive drugs such as systemic cortico-steroids.12 Demirçay et al12 described a patient with skin lesions that rapidly cleared after the administration of oral methylprednisolone (48 mg/d) and oral dapsone (100 mg/d). The methylprednisolone and dapsone were withdrawn after 12 and 16 weeks, respectively. There was no recurrence during the 1-year follow-up period.12Kolb-Mäurer et al19 described a patient who was treated with pulsed intravenous corticosteroids and continued to develop new papular and vesicular skin lesions. However, when oral acitretin was added to the patient’s regimen, the skin lesions cleared.19 There are several case reports of the successful use of hydroxychloroquine in LP.20,21
Cutaneous, nail, and oral LP also can be treated with TNF-α inhibitors (eg, adalimumab, etanercept) with resolution of lesions.22-25 However, we have not been able to find any reports of treating LPP with biologic medications in a search of PubMed articles indexed for MEDLINE using the terms lichen planus pemphigoides and biologic treatments/therapies. Given the fact that TNF-α and other inflammatory cytokines are involved in the pathogenesis of BP and LP, it is feasible that they also may be involved in the pathogenesis of LPP.
In our patient with cutaneous LPP, we chose to use ustekinumab instead of a primary TNF-α inhibitor because ustekinumab indirectly blocks TNF-α, as well as other proinflammatory cytokines such as IFN-γ, IL-17, and IL-22, which also could have played a role in the patient’s disease. Our goal was to use ustekinumab as a potential corticosteroid-sparing agent. Ustekinumab greatly improved her skin condition and allowed us to discontinue other medications.