Recently, smoking has been implicated as a potential risk factor in the disease.3,15 Certain environmental exposures have also been recognized as a possible risk factor. In Guam, an ALS-like syndrome has been identified among members of the Chamorro tribe. This syndrome has been linked to a neurotoxin in the seed of the cycad nut, a tropical plant endemic to the area, which was used in the 1950s and 1960s in the human food supply.3,16
ETIOLOGY
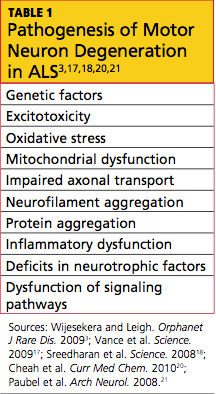
Familial ALS is a genetically transmitted degenerative disease. Twenty percent of cases involve the long arm of chromosome 21, which is responsible for coding of superoxide dismutase (SOD1). Mutations in SOD1, an RNA-processing protein called FUS, and the DNA-binding protein TDP-43, have all been identified in cases of familial ALS.17,18
The exact etiology of sporadic ALS remains unknown, but gene mutations have also been implicated, including the ANG gene21 and TDP-43.18 Other theories include increased levels of glutamate (which have been detected in cerebrospinal fluid and serum of patients with ALS), mitochondrial dysfunction, free radical injury, programmed cell death, neurofilament defects, viral infections, and autoimmune dysfunction19,20 (see Table 13,17,18,20,21). It is possible that a combination of factors is involved in the development of sporadic ALS.
PATHOPHYSIOLOGY
The pathophysiology of ALS involves degeneration of the UMN and LMN axons, which leads to glial scarring and possible impairment of the glial cells’ ability to store excess glutamate.22 ALS affects the central nervous system, specifically the anterior horn cells in the spinal cord and the cranial nerve nuclei (X, XI, XII) of the LMNs, and the corticospinal tract and corticobulbar pathway of the UMNs. Bulbar and limb muscles innervated by LMNs are subject to atrophy, whereas cognition, coordination, sensation, the oculomotor system, and sphincters are typically spared.3
CLINICAL FEATURES
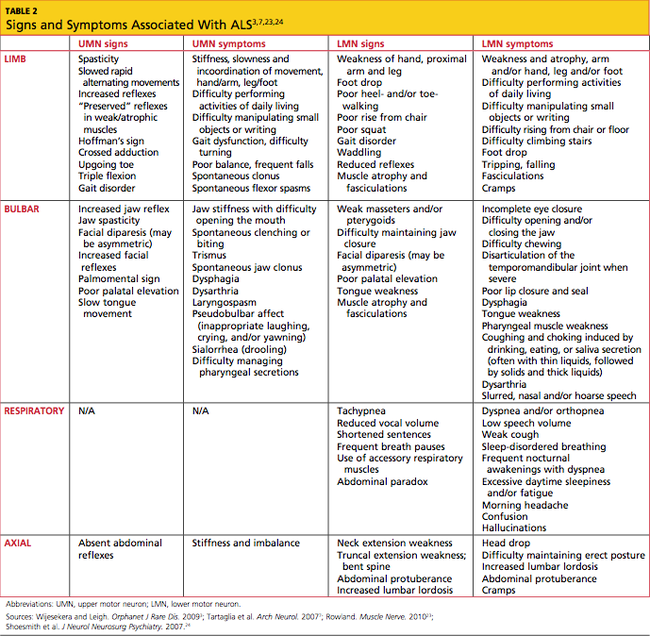
In most patients, ALS symptoms characterize either limb onset or bulbar onset (see Table 23,7,23,24), with limb onset being the more common (about 75% vs about 25% of cases, respectively).1 Typically, patients with limb-onset ALS complain of rapidly progressive, asymmetric weakness in an extremity, followed by focal muscle atrophy with cramping and fasciculations, and eventually, spasticity. Weakness generally begins in one hand, arm, foot, or leg. Patients may notice increased episodes of tripping, clumsiness when they run or walk, a “dropped foot” gait, and/or a decline in manual dexterity.25
The weakness often develops insidiously; patients may notice that symptoms are exacerbated by cold weather.3 Eventually, the bulbar muscles are affected, resulting in dysphagia, dysarthria, and dysphasia. Occasionally, patients encounter bladder dysfunction (urgent micturition), sensory symptoms, and cognitive symptoms (eg, dementia, parkinsonism).26 Multisystem involvement is possible. Ultimately, respiratory compromise or other pulmonary complications ensue, representing a primary cause of mortality in ALS patients.3,24
In bulbar-onset ALS, patients first notice symptoms of dysphasia and dysphagia. They may complain of slurred speech, nasal or low-volume speech, and/or inhibited tongue mobility. The risk for aspiration is increased. The majority of patients with bulbar-onset ALS experience sialorrhea (excessive drooling) because they have difficulty swallowing their saliva. In most patients, mild UMN-type bilateral facial weakness affects the lower half of the face.3 As is the case with limb-onset ALS, bulbar-onset ALS progresses to respiratory compromise.27
In less than 3% of patients with ALS, presentation begins with respiratory weakness and no significant limb or bulbar symptoms.24,28 Patients with respiratory-onset ALS experience symptoms associated with nocturnal hypoventilation, including daytime hypersomnolence, morning headaches, impaired concentration, irritability, anorexia, mood changes, dyspnea, orthopnea, and disturbed sleep; or they may experience type 2 respiratory failure.28,29
Patients with axial symptoms of ALS present with neck weakness and may complain of posterior neck pain or strain with a gradually worsening tendency of the head to tip forward. These patients often support the chin with one hand. Those with axial truncal weakness often complain of difficulty maintaining erect posture when standing and of stooping as they walk. Some patients support the trunk by placing their hands in their front pants pockets or on their upper thighs. They may report some relief when pushing a grocery cart.7,23,24
Symptoms of ALS can be present for weeks or months before a patient consults with a health care provider. The average time span from onset of initial symptoms to diagnosis of ALS is about one year.1 Due to the unpredictable pattern of progression and variability of symptoms among patients, it is difficult to approximate a time frame for symptom progression; for some patients, the disease progresses slowly, while others deteriorate rapidly.30
PHYSICAL EXAMINATION FINDINGS
At onset, the typical presentation of ALS includes muscle weakness in one limb as well as visible fasciculations. As the disease progresses, focal wasting of muscle groups occurs in all four extremities. Particularly involved are the muscles of the hands, forearms, or shoulders in the upper limbs; and of the proximal thigh or distal foot muscles in the lower limbs.31 Deep tendon reflexes are symmetrically brisk. Spasticity, evident in the upper limbs, may present as increased tone.32