In patients with bulbar dysfunction, dysarthria may arise from either LMN pathology or pseudobulbar palsy caused by a UMN disorder, leading to slow, slurred speech or speech with a nasal quality. Tongue fasciculations will be present, as will atrophy and diminished mobility of the tongue.33 The gag reflex remains intact, even brisk, but weakness may occur in the muscles of the soft palate.3 Facial weakness is sometimes seen late in the disease, as evidenced by difficulty sealing the lips or puffing out the cheeks. The jaw jerk will be brisk, indicating that cranial nerve V is intact.34
A pseudobulbar affect, which is best described as emotional lability, may be present. The patient may have a history of exaggerated expression of emotion, such as uncontrollable crying, laughing, or both.35 Cognition, coordination, sensation, the oculomotor system, and sphincters are generally spared. However, cases of frontotemporal dementias coexisting with ALS have been reported; affected patients exhibit cognitive impairment, compulsive behaviors, and personality changes, and they may experience shorter survival.4,24,36
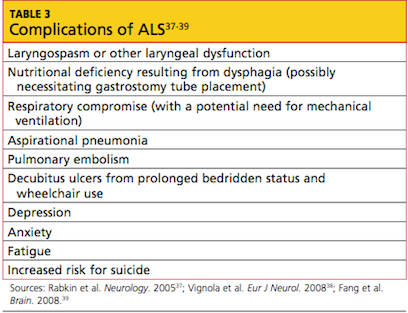
Evidence of known complications of ALS may also be noted during the physical examination; see Table 3.37-39
DIAGNOSIS OF ALS
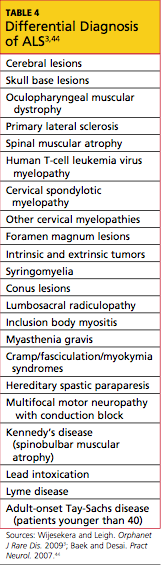
In current research, use of structural MRI, magnetic resonance spectroscopy, and diffusion tensor imaging is being examined to detect thinning in the primary motor cortex, fractional anisotropy in the corpus callosum, patterns of gray and white matter atrophy, and other proposed diagnostic markers for ALS.40-43 However, no single specific diagnostic test has yet been proven to identify ALS; rather, it remains a disease of exclusion (see Table 43,44). For a confirmed diagnosis of ALS to be made, the patient must display:
Evidence of LMN degeneration as found through clinical, neuropathologic, or electrophysiological examination
UMN degeneration detected by clinical examination, and
Progressive spread of signs or symptoms within a region or to other regions.3,45-47
Other disease processes that might explain the signs of UMN and/or LMN degeneration must be excluded, such as cervical spinal disease, myasthenia gravis, multifocal motor neuropathy, lead intoxication, and Lyme disease.45
Imaging studies are not required in ALS cases that have clinically definite disease with bulbar or pseudobulbar onset.47 Otherwise, the essential role of neuroimaging is to exclude a treatable structural lesion that may mimic ALS by producing UMN and LMN signs in varying degrees.48
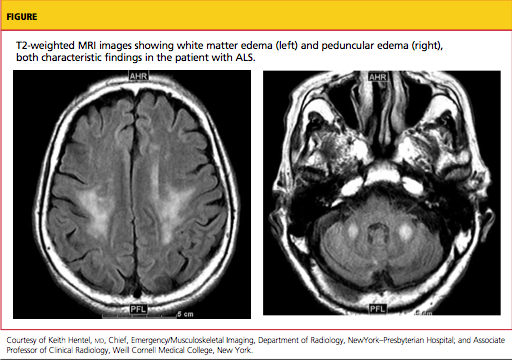
Typically, MRI of the head and spine is ordered in patients with suspected ALS (see figure); MRI can reveal lesions in the corticospinal tracts that occur in ALS. The most characteristic finding on T2-weighted MRI is hyperintensity of the corticospinal tracts, which is visualized best in the brain and brainstem, and to a lesser extent in the spinal cord.49 Decreased signal intensity in the motor cortex has been reported on MRI in cases of ALS.50
Additional diagnostic procedures that are useful in excluding other disease processes include blood and cerebrospinal fluid (CSF) samples,3,44 four-limb electromyography (EMG),44 nerve conduction studies, motor unit number estimations, and muscle biopsies.47
Autopsy results of patients with ALS demonstrate:
Neuron loss, especially in lumbar and cervical enlargements
Nonexistent or atrophic neurons in the motor nuclei of the pons and medulla, and in the anterior horn cells of the spinal cord
Degeneration of the lateral columns of the spinal cord, and
Atrophy of the ventral roots.31,51
PROGNOSIS
The overall five-year survival rate for patients with ALS has been reported between 7% and 14%,52,53 and the mortality rate rises in patients older than 75 and in those with bulbar signs.30 The rate of disease progression varies greatly among ALS patients and may be hastened with advancing age, female gender, presence of bulbar features, and absence of a significant other.37,53,54
The average life span for a patient with limb-onset ALS is two to five years from diagnosis, whereas patients affected by the bulbar form usually succumb within six to 18 months.30,53 In patients who present with respiratory symptoms, Shoesmith et al24 have reported a mean time of 14.9 months from initial symptoms to need for full-time ventilation, and of 27.0 months from symptom onset to death.
Advance directives, end-of-life care, and respiratory and nutritional management become essential issues during late stages of ALS; thus, they should be discussed with patients and their relatives at the time of diagnosis or shortly thereafter.
TREATMENT
Pharmacologic Options
There are currently no treatments to halt the progression of ALS or to reverse the disease process. Riluzole, currently the only FDA-approved drug for treatment of ALS, has been shown to slow the loss of muscle strength and to prolong life by an average of two to three months.55 Riluzole targets and blocks glutamate transporters on the presynaptic neuron, decreasing glutamate release and reducing excitotoxicity20 (ie, overstimulation of the postsynaptic receptors). These effects support the theory that ALS may result from excess glutamate and help to explain the increased levels of glutamate found in the serum and CSF of ALS patients.19