Twice as common as autism and half as well-known,1 autosomal polycystic kidney disease (ADPKD) occurs in one in 400 to one in 1,000 people.2 It is an inherited progressive genetic disorder that causes hypertension and decreased renal function and, over time, can lead to kidney failure. Two polycstin genes that code for ADPKD, PKD1 and PKD2, were identified in 1994 and 1996, respectively.3,4 Awareness and understanding of the genes responsible for ADPKD have increased clinicians’ ability to identify at-risk patients and to slow or alter the course of the disease.
Case Presentation
A 45-year-old black man presents to your office with severe, nonradiating back pain and new-onset hypertension. Regarding the pain, he stated, “I turned around to see who kicked me, but no one was there.” When the pain began, he went to see the nurse at the school where he is employed, and she found that his blood pressure was high at 162/90 mm Hg. Although the patient’s back pain is resolving, he is very concerned about his blood pressure, since he has never had a high reading before.
He is the baseball coach and physical education teacher at the local high school and is in excellent physical condition as a result of his professional interaction with teenagers every day. He does not smoke or use any illicit drugs but does admit to occasional alcohol consumption. His medical history is significant only for occasional broken fingers and twisted ankles, all occurring while he was engaged in sports.
His family history includes one brother without medical problems, a brother and a sister with hypertension, a sister with diabetes and obesity, and a brother with a congenital abnormality that required a living donor kidney transplant at age 17 (the father served as donor). No family-wide workup has ever been done because no one practitioner has ever made a connection among these conditions and considered a diagnosis of ADPKD.
The patient’s blood pressure in the office is 172/92 mm Hg while sitting and 166/88 mm Hg while standing. He is somewhat sore with a localized spasm in the lumbar-sacral area but no radiation of pain. The patient has trouble touching his toes but reports that he can never touch his toes. His straight leg lift is negative. The rest of his physical exam is noncontributory.
What should be the next step in this patient’s workup?
PATHOPHYSIOLOGY
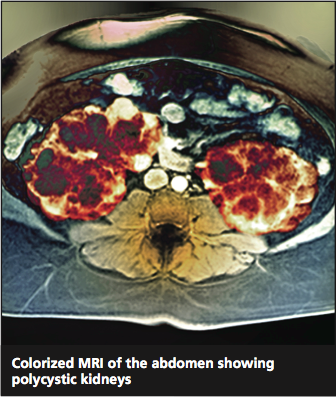
ADPKD is a progressive expansion of numerous fluid-filled cysts that result in massive enlargement of the kidneys.5 Less than 5% of all nephrons become cystic; however, the average volume of a polycystic kidney is 1,000 mL (normal, 300 mL), that is, the volume of a standard-sized pineapple. Even with this significant enlargement, a decline in the glomerular filtration rate (GFR) is not usually seen initially. Each cyst is derived from a single hyperproliferative epithelial cell. Increased cellular proliferation, followed by fluid secretion and alterations in the extracellular matrix, cause an outpouching from the parent nephron, which eventually detaches from the parent nephron and continues to enlarge and autonomously secrete fluid.6,7
PKD1 and PKD2 are two genes responsible for ADPKD that have been isolated so far. Since there are families carrying neither the PKD1 nor the PKD2 gene that still have an inherited type of ADPKD, there is suspicion that at least one more PKD gene, not yet isolated, exists.8 It is also possible that other genetic or environmental factors may be at play.9,10
In 1994, the PKD1 gene was isolated on chromosome 16,3 and it was found to code for polycystin 1. A lack of polycystin 1 causes an abnormality in the Na+/K(+)-ATPase pumps, leading to abnormal sodium reabsorption.11 How and why this happens is not quite clear. However, the hypertension that is a key objective finding in patients with ADPKD is thought to result from this pump abnormality.
PKD2 is found on the long arm of chromosome 4 and codes for polycystin 2.4 Polycystin 2 is an amino acid that is responsible for voltage-activated cellular calcium channels,5 again explaining the hypertension so commonly seen in the course of ADPKD. ADPKD-associated hypertension may be present as early as the teenage years.12
EPIDEMIOLOGY
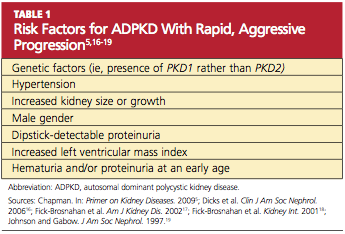
More than 85% of ADPKD cases are associated with PKD1, and this form is called polycystic kidney disease 1 (PKD 1), the more aggressive form of the disease.13,14 PKD 2 (the form associated with the gene PKD2), though less common, is also likely to progress to end-stage renal disease (ESRD), but at a later age (median age of 74 years, compared with 54 in patients with PKD 1).14 ADPKD accounts for about 5% of cases of ESRD in North America,9 but for most patients, presentation and decreased renal function do not occur until the 40s.15 However, patients with the risk factors listed in Table 15,16-19 are likely to experience a more rapid and aggressive form of the disease.