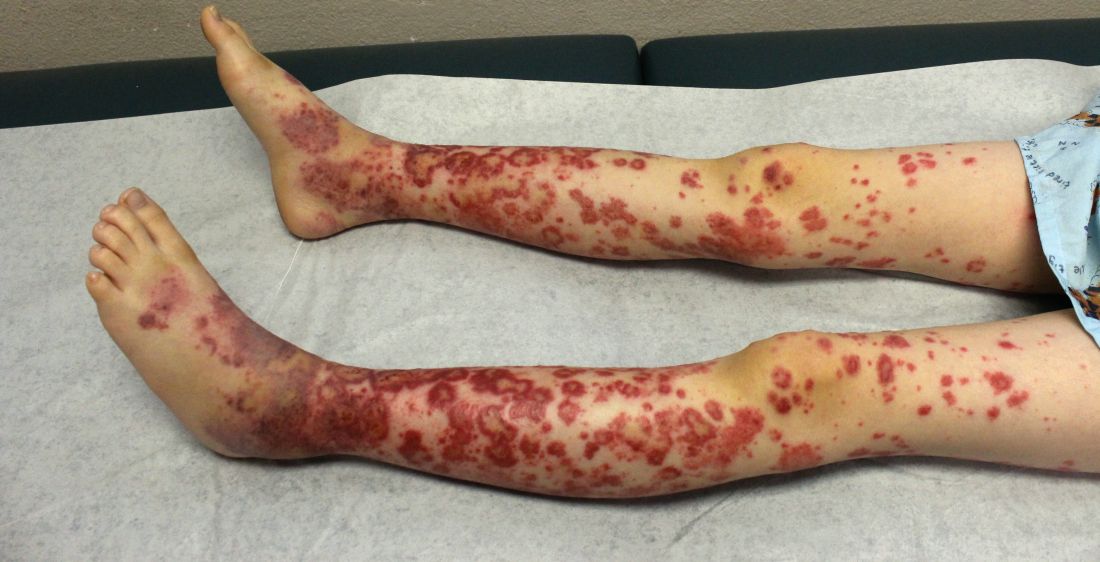
The patient was diagnosed with Henoch-Schönlein purpura (HSP) based on clinical presentation of the lesions and associated symptoms of arthralgia and abdominal pain. Urinalysis was obtained and found to be unremarkable, at presentation and follow-up, and treatment with naproxen 5 mg/kg divided into two doses per day was started for pain relief. A prednisone taper starting at 1 mg/kg per day for 3 weeks also was started due to the presence of severe abdominal pain and bullae on exam. The patient was followed with regular urine studies and blood pressure checks for 2 months, and these also were within normal limits.
HSP, also known as anaphylactoid purpura and immunoglobulin A (IgA) vasculitis, is a small vessel leukocytoclastic vasculitis characterized by the perivascular deposition of IgA1-based immune complexes in the walls of arterioles and postcapillary venules.1 In the vast majority of cases, the condition resolves spontaneously in 4-6 weeks and does not require any specific treatment,2 although NSAIDs and systemic corticosteroids can be used for mild-to-moderate and severe pain, respectively.3
HSP is the most common vasculitis in children, with a peak incidence in boys under the age of 5 years. It occurs worldwide, more commonly among whites and Asians, less commonly among blacks, and recent studies from the Czech Republic,4 Taiwan,5 Spain,6 France,7 South Korea,8 and the United Kingdom9 have shown similar incidence rates of 10-20 per 100,000 children. HSP does occur in adults, but is less common, and is known to carry a worse prognosis – in particular, a higher risk of progression to chronic kidney disease. The disease is more commonly seen in winter months,1 unsurprisingly as upper respiratory tract infections also are more common in these months.10
Pathogenesis
The exact pathogenesis of HSP is the subject of ongoing investigation and continued controversy. Mutations and polymorphisms in mannose-binding lectin, interleukins 1 and 8, vascular endothelial growth factor, and alpha-1-antitrypsin have been associated with HSP.3 Immunoglobulin A (IgA) normally exists in two heavily glycosylated forms – IgA1 and IgA2. Abnormal glycosylation, particularly undergalactosylation, of IgA1, the predominant form of IgA in serum and mucosal secretions, has been linked to HSP.11 HSP has been associated with group A streptococcal infections, Bartonella henselae (cat scratch fever) and numerous drugs,12 although no definitive causal or mechanistic explanation has been identified.
Diagnosis
Two major diagnostic criteria for HSP are widely in use, one developed by the American College of Rheumatology (ACR) in 199013 and the other by the European League Against Rheumatism (EULAR) in 2005.14 Both the ACR and EULAR criteria include acute abdominal pain, purpura, and microscopic evidence of vasculitis. Almost all patients with HSP have cutaneous purpura, and many of these patients have palpable purpura, which is pathognomonic of a leukocytoclastic vasculitis, but palpable purpura is not needed for diagnosis. The ACR criteria additionally include age of 20 years or younger, while the EULAR criteria include arthralgias and the presence of hematuria or proteinuria. Ancillary testing usually is not required to make the diagnosis, but when the diagnosis is not clear histopathologic analysis of a skin sample can identify leukocytoclastic vasculitis. Other laboratory studies that may be needed to rule out other conditions, as well as other organ involvement, include a complete blood count, which can be done to rule out thrombocytopenia as a cause of purpura, a metabolic panel, coagulation studies, occult blood test of stool, abdominal imaging, and urinalysis (UA), which can identify proteinuria or hematuria.
Abdominal pain in HSP is believed to be a result of vasculitis of the gastric, mesenteric, and/or colic vasculature. Bleeding from the inflamed vasculature rarely can lead to gross hematochezia, frank melena, or hematemesis. One serious, potential complication of HSP-related mesenteric vasculitis is intussusception, which is otherwise rare in children older than 2 years. Intussusception should be suspected if features of the classic triad of episodic abdominal pain, sausage-shaped abdominal mass, and currant jelly stool are present. Abdominal ultrasound can help to determine whether intussusception is present.
The purpura in HSP presents in waves or crops, and crops last 5-10 days each. Complete resolution takes 4-6 weeks. If biopsy is desired to confirm the diagnosis, it should be done on a lesion less than 24 hours old. This allows for identification of perivascular IgA on histopathology: beyond 24 hours, IgG and IgM also leak out, contributing to a less specific histopathologic picture.
Accurate diagnosis of HSP is important to guide therapy and anticipate potential complications. Wegener’s granulomatosis (A), also known as granulomatosis with polyangiitis, classically involves the upper and lower respiratory tract and the kidneys, leading to a presentation of epistaxis, cough, and hypertension. It occurs more commonly in adults than children. Finkelstein disease (B), also known as acute hemorrhagic edema of infancy (AHEI), is characterized by the development of petechial, urticarial, or targetoid plaques over 24-48 hours with tender edema and fever in children aged less than 2 years. Unlike HSP, AHEI typically does not involve the gastrointestinal tract, kidneys, or joints. Biopsy of skin lesions of AHEI reveals IgA deposition and leukocytoclastic vasculitis, leading some authors to consider it a closely related entity to HSP. Microscopic polyangiitis (D) is an uncommon pauci-immune vasculitis similar to Wegener’s granulomatosis, but lacking granulomas. It presents typically in the 5th decade of life with fever, fatigue, weight loss, and renal involvement. IgA nephropathy (E), also known as synpharyngitic nephritis and Berger disease, is less likely than HSP to cause a rash, joint pain, or abdominal pain. The nomenclature of HSP (whose alternate name is IgA vasculitis) reflects the multi-organ nature of HSP in comparison to IgA nephropathy, which is more likely to be limited to the kidneys.
Treatment
Aside from intussusception and renal disease, which may result from HSP, treatment is not typically required for HSP as it resolves spontaneously. Patients with significant arthralgias are likely to benefit from NSAIDs such as naproxen 5-20 mg/kg per day, although NSAIDs should be avoided if there is significant renal dysfunction or GI bleeding. Patients with severe abdominal pain or joint pain may be more likely to benefit from oral corticosteroids, particularly prednisone 1-2 mg/kg per day. A meta-analysis showed that corticosteroids significantly reduce the duration of symptoms if given early in the course of disease.15
The prognosis is usually excellent, except for a very small sample of the population (5%) that can develop end-stage renal disease. It is recommended that all children with HSP continue monitoring blood pressure and UA either weekly or biweekly for the first 2 months and then once a month for 6-12 months.16
First described in 1801 by a British physician, HSP is a common and usually self-limited disease for which our understanding has advanced greatly over the past 2 centuries, yet for which many important questions regarding pathophysiology remain unanswered. No diagnostic tests or treatments are needed for the majority of patients. Providers should include HSP in the differential diagnosis for the child with unexplained abdominal pain, renal dysfunction, or nonthrombocytopenic purpura.
Mr. Kusari is a medical student at the University of California, San Diego. Dr. Matiz is a practicing dermatologist at Southern California Permanente Medical Group in La Mesa, California. Dr. Matiz and Mr. Kusari said they had no relevant financial disclosures. Email them at pdnews@frontlinemedcom.com.
References
1. “Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence”, 5th ed. (New York: Elsevier, 2016).
2. Lancet. 2007;369(9566):976-8.
3. “Dermatology”, 3rd ed. (Philadelphia: Elsevier Saunders, 2012).
4. J Rheumatol. 2004 Nov;31(11):2295-9.
5. Rheumatology (Oxford). 2005 May;44(5):618-22.
6. Medicine (Baltimore). 2014 Mar;93(2):106-13.
7. Rheumatology (Oxford). 2017;56(8):1358-66.
8. J Korean Med Sci. 2014 Feb;29(2):198-203.
9. Lancet. 2002 Oct 19;360(9341):1197-202.
10. Rhinology. 2015 Jun;53(2):99-106.
11. PLoS One. 2016 Nov 21;11(11):e0166700.
12. Pediatr Infect Dis J. 2002 Jan;21(1):28-31.
13. Arthritis Rheum. 1990 Aug;33(8):1114-21.
14. Ann Rheum Dis. 2006 Jul;65(7):936-41.